This Week on Purna AI Molecular Intelligence Platform

Two things shipped on MIP this week. One helps researchers stop context-switching into BioRender every time they need a figure. The other brings protein language models directly into the chat, for both generic proteins and paired antibodies. Both are available today to every MIP user.
This post walks through what each capability does, how to reach it, and the kinds of workflows it unlocks.
Scientific figures, on demand
MIP can now draw publication-quality scientific figures from a natural-language prompt, inline in the chat. Diagrams, pathways, schematics, illustrations, and multi-panel infographics all render in place, alongside the analysis that inspired them.
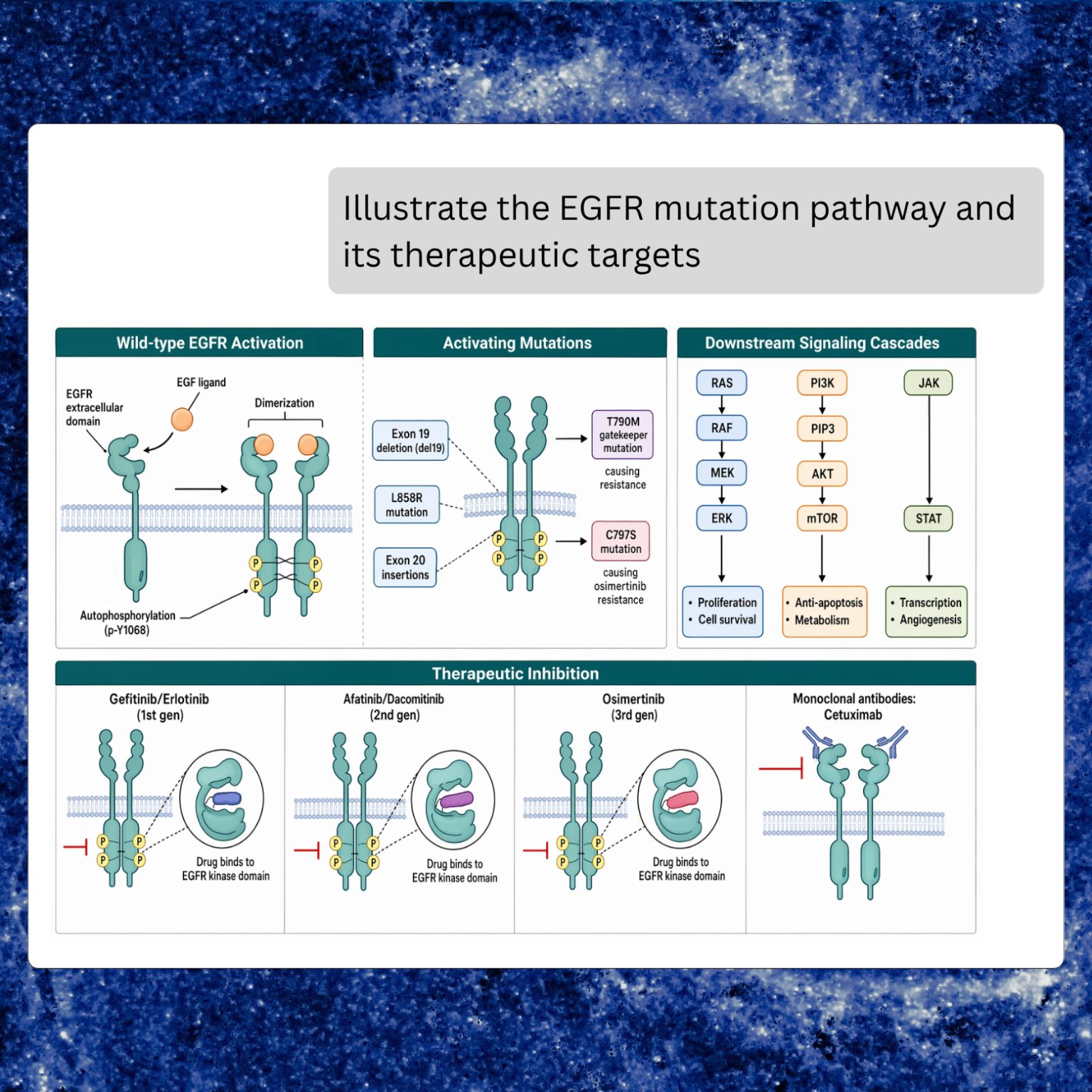
The prompt-to-figure loop is the point. A researcher can ask a question, let MIP pull evidence from literature and databases, and then ask for a figure built from that same context in the next turn. Labels, residue identifiers, gene names, and pathway members that were just mentioned in the conversation are carried into the drawing, rather than re-specified from scratch.
Typical prompts:
- “Draw the MAPK/ERK signaling cascade with the key kinases labeled.”
- “Illustrate antibody-antigen binding at the CDR loops.”
- “Create a three-panel infographic of the workflow we just ran.”
- “Make a schematic of the PCSK9 mechanism, based on the papers you just found.”
A few practical notes. Any label you want rendered verbatim should be quoted in double quotes, which MIP passes through unchanged. Intent options (diagram, pathway, schematic, infographic, illustration) nudge the output toward a specific figure style, and in most cases the default picks the right one. Size and quality default to sensible values but can be set for posters, slide headers, or draft iterations.
The generated figure is a first draft meant to save the BioRender hour, not a final regulatory-grade deliverable. For papers and clinical reports, review and polish in a dedicated tool before publishing. For everything else — internal decks, exploratory notes, figures for a draft manuscript — the inline output is usually good enough on the first or second iteration.
Full documentation: Scientific Figures.
Protein embeddings and mutation likelihoods
MIP now runs protein language models directly in the chat, for two distinct use cases:
- ESM-2 on generic proteins (enzymes, receptors, scaffolds, any non-antibody protein)
- AbLang-2 on paired antibodies (heavy plus light chain)
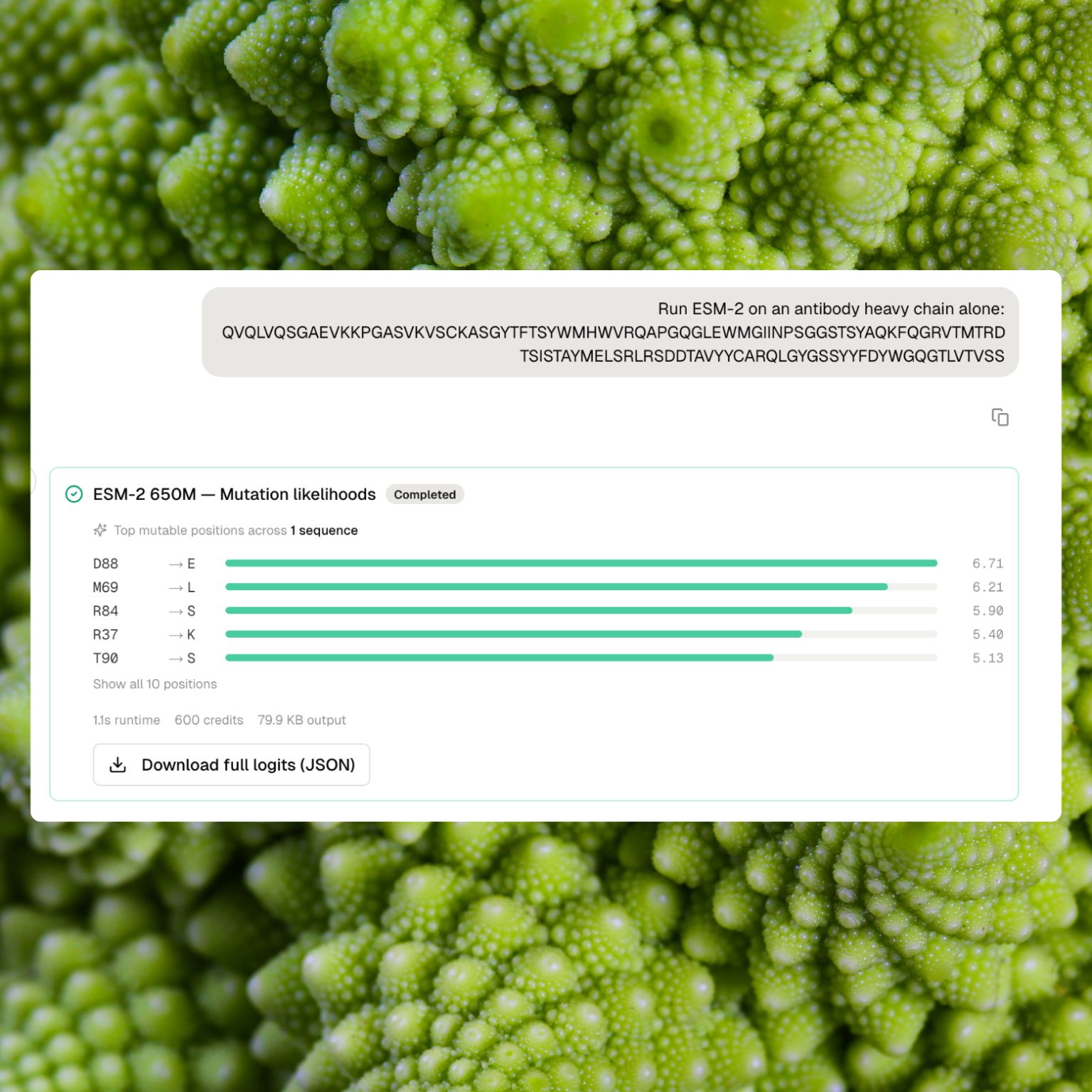
Each model family supports two actions: embeddings (a fixed-length numerical representation of a full sequence) and mutation likelihoods (a per-position score for every possible amino acid substitution).
Embeddings
Embeddings summarise a sequence as a single vector. Two sequences with similar embeddings are treated as similar by the underlying model, typically reflecting similar fold, family, or function. The practical uses are straightforward: cluster a set of candidates, search a library for near-neighbours, compare two proteins numerically, or feed the vectors into a downstream property-prediction model.
A researcher can submit up to eight sequences (ESM-2) or up to thirty-two paired antibodies (AbLang-2) in a single run. The result card returns a compact summary with the sequence count and the vector dimensionality. The full numerical matrix is saved as a downloadable JSON dataset, ready to load into a notebook or a clustering pipeline.
Mutation likelihoods
Mutation likelihoods rank every possible single-residue substitution at every position along a sequence. High scores at a position signal plausible alternatives to the natural residue, which is often a useful proxy for evolutionary tolerance, flexibility, or an engineerable site. Low scores signal strong preference for the observed residue.
The result card surfaces the top mutable positions with their highest-likelihood substitutions, in a compact list. The full per-position by twenty-amino-acid matrix goes to the dataset for deeper inspection.
The intended workflow: embed first to pick representatives, then score mutation likelihoods on those representatives to find tolerant positions, then design a small experimental set around the top-scoring residues. Pair with structure prediction to confirm that top-scoring mutations sit in plausible structural contexts before committing them to the wet lab.
When to pick ESM-2 vs AbLang-2
Use AbLang-2 for antibodies. It is trained on paired antibody repertoires, so its likelihoods reflect CDR variability, framework conservation, and the statistics of natural heavy-light pairings. Use ESM-2 for everything else. It is a general-protein model with broader coverage and handles sequences up to 2,048 residues.
A single workspace can have any combination of the four capabilities enabled. Teams that only work with antibodies can turn off the ESM-2 entries and keep the chat focused; teams working on enzymes and antibodies alike can enable all four.
Pricing
Both model families bill on actual compute time, at ten credits per second rounded up to the nearest minute. Most runs complete in under a minute. Identical inputs within ten minutes return the cached result at no extra cost.
Full documentation: Protein Embeddings & Mutation Likelihoods.
Where this fits in MIP
Both capabilities extend the pattern MIP has been building toward: the analysis steps that follow a question happen in the same environment as the question itself, with tool outputs flowing directly into each other. A researcher can decompose an antibody engineering problem across several turns in a single chat:
- Ask AbLang-2 for mutation likelihoods on the reference antibody.
- Inspect the top mutable CDR positions in the result card.
- Predict the 3D structure of the parental antibody to check the structural context.
- Ask for a figure summarising the proposed mutations against the structure, for a slide or a manuscript draft.
The same loop works for a generic protein, with ESM-2 standing in for AbLang-2. None of these steps require opening a separate tool, a separate browser tab, or a separate notebook. The outputs are carried forward in the conversation and available in the Files view.
Getting started
Both features are live in MIP today. Scientific figure generation is enabled by default. Protein embeddings and mutation likelihoods are managed per model in capability settings, so users can enable only the ones they need (AbLang-2 Embeddings, AbLang-2 Mutation likelihoods, ESM-2 Embeddings, ESM-2 Mutation likelihoods).
For researchers new to the platform, up to ten thousand dollars in MIP research credits are available through our research credits program. Existing users can start using both capabilities from their next chat.
More updates next week.
Purna AI’s Molecular Intelligence Platform (MIP) is an AI-powered workspace for biology teams. It brings together molecular analysis, variant interpretation, protein structure prediction, de novo enzyme design, and clinical database integrations into one environment. Built for teams who work with biological data and need consistent, reproducible answers without juggling disconnected tools. Learn more at purna.ai.
Explore Purna's Molecular Intelligence Platform
AI-powered workspace for biology teams to accelerate drug discovery from target identification to lead optimization.
Try Purna AI →
