How to Predict the Impact of Protein Mutations on Structure and Function

A single amino acid change can disable an enzyme, destabilize a protein fold, or leave function entirely intact. The challenge for researchers and clinicians is telling the difference. With over 4 million missense variants catalogued in human populations, and most classified as variants of uncertain significance (VUS), predicting the impact of protein mutations on structure and function has become one of the most consequential problems in molecular biology.
This guide walks through a practical workflow for protein mutation impact prediction: from retrieving a structure, to visualizing the mutation site, to running stability and conservation analyses, to interpreting the results. Every step can be done computationally, and platforms built around molecular intelligence now integrate these steps into a single workspace.
Why Protein Mutation Impact Prediction Matters
Not all mutations are created equal. A substitution buried in a flexible loop may have no measurable effect, while the same change in a catalytic active site can abolish function entirely. Understanding this requires reasoning about protein structure, not just sequence.
The clinical stakes
In clinical genomics, the American College of Medical Genetics and Genomics (ACMG/AMP) framework uses protein-level evidence as part of variant classification. Criteria like PM1 (located in a mutational hotspot or functional domain), PP3 (computational evidence supports a deleterious effect), and PM5 (novel missense at a position where a different pathogenic missense has been seen) all depend on understanding how a mutation affects the protein.
Yet most clinical labs still classify the majority of missense variants as VUS. In ClinVar, roughly 50% of all submitted variants carry this label. Resolving them faster requires better computational tools for predicting mutation impact, tools that combine structural, evolutionary, and functional evidence.
The research applications
Beyond clinical genetics, protein mutation impact prediction is central to protein engineering, drug target validation, and understanding disease mechanisms. Researchers designing therapeutic proteins need to predict which mutations will improve stability or binding affinity. Teams studying disease mechanisms need to understand which mutations in a gene of interest actually disrupt function versus those that are tolerated.
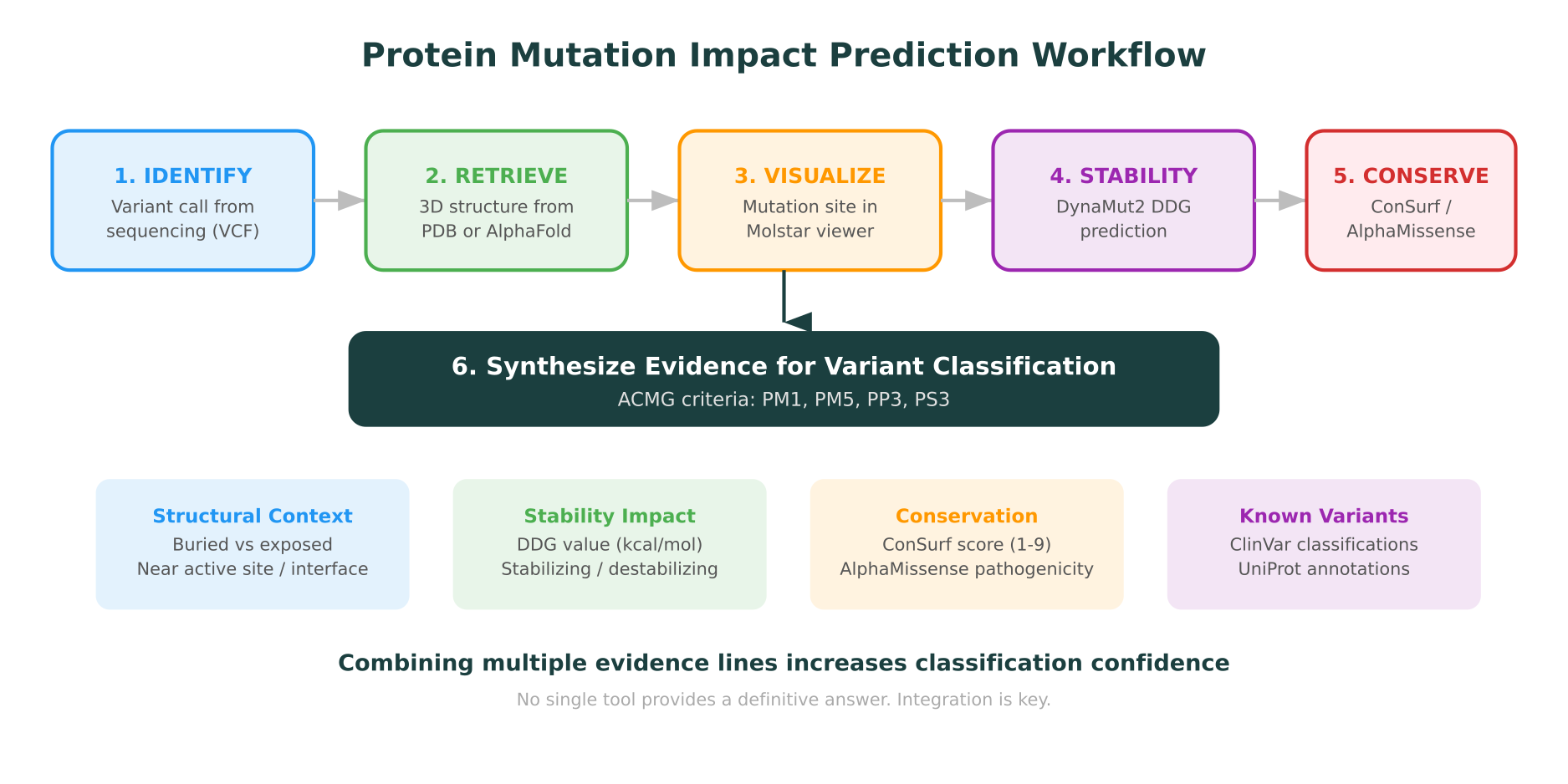
Step 1: Retrieve the Protein Structure
The foundation of any structural mutation analysis is having a reliable 3D model of the protein.
Experimental structures from the Protein Data Bank
The RCSB Protein Data Bank (PDB) contains over 220,000 experimentally determined structures as of early 2026, solved by X-ray crystallography, cryo-EM, and NMR spectroscopy. If your protein of interest has an experimental structure, this is the gold standard starting point.
Key considerations when selecting a PDB structure:
- Resolution matters. For mutation analysis, structures below 2.5 angstrom resolution are preferred. Lower resolution structures may have poorly resolved side chains, which undermines mutation impact analysis.
- Coverage matters. Many PDB entries cover only a domain or fragment. Check that the mutation site falls within the resolved region.
- Biological assembly matters. Proteins often function as multimers. A mutation at a subunit interface may look benign in an isolated chain but could disrupt dimerization.
Predicted structures from AlphaFold
For proteins without experimental structures, the AlphaFold Protein Structure Database provides predicted models for over 214 million proteins, covering nearly every known protein sequence. AlphaFold 3, published in Nature in May 2024, extended predictions to protein complexes with DNA, RNA, ligands, and ions.
When using AlphaFold models, pay attention to the per-residue confidence score (pLDDT):
- pLDDT > 90: High confidence. Reliable for mutation analysis.
- pLDDT 70-90: Moderate confidence. Backbone is likely correct, but side-chain positioning may be approximate.
- pLDDT < 70: Low confidence. Often corresponds to disordered regions. Structural mutation analysis in these regions is unreliable.
AlphaFold models are excellent for assessing whether a mutation falls in a structured domain, but they represent a single static conformation. They don’t capture protein dynamics, allosteric effects, or the impact of post-translational modifications.
Step 2: Visualize the Mutation Site
Before running any computational analysis, visualizing the mutation in its structural context provides immediate insight.
Molstar: the modern standard
Molstar is an open-source, web-based molecular visualization tool that has become the default viewer for the RCSB PDB and the AlphaFold database. Unlike desktop tools like PyMOL or ChimeraX, Molstar runs entirely in the browser with no installation required.
When visualizing a mutation site, look for:
- Burial vs. exposure. Is the residue buried in the hydrophobic core or exposed on the surface? Buried residues are far more sensitive to substitutions, especially changes in size or charge.
- Secondary structure context. Is the residue in an alpha helix, beta sheet, or loop? Proline substitutions in helices are typically disruptive because proline breaks helical geometry.
- Nearby functional features. Is the residue near the active site, a binding interface, a disulfide bond, or a known post-translational modification site?
- Hydrogen bonding network. Does the wild-type residue participate in hydrogen bonds that would be broken by the substitution?
Platforms like Purna AI’s MIP integrate Molstar directly into the analysis workspace, so researchers can visualize the structure, check the mutation context, and run stability analysis without switching tools.
Step 3: Assess Structural Stability with DynaMut2
Protein stability is one of the most informative signals for predicting whether a mutation is tolerated or damaging. DynaMut2 is a widely used tool for predicting changes in protein stability and dynamics upon mutation.
What DynaMut2 measures
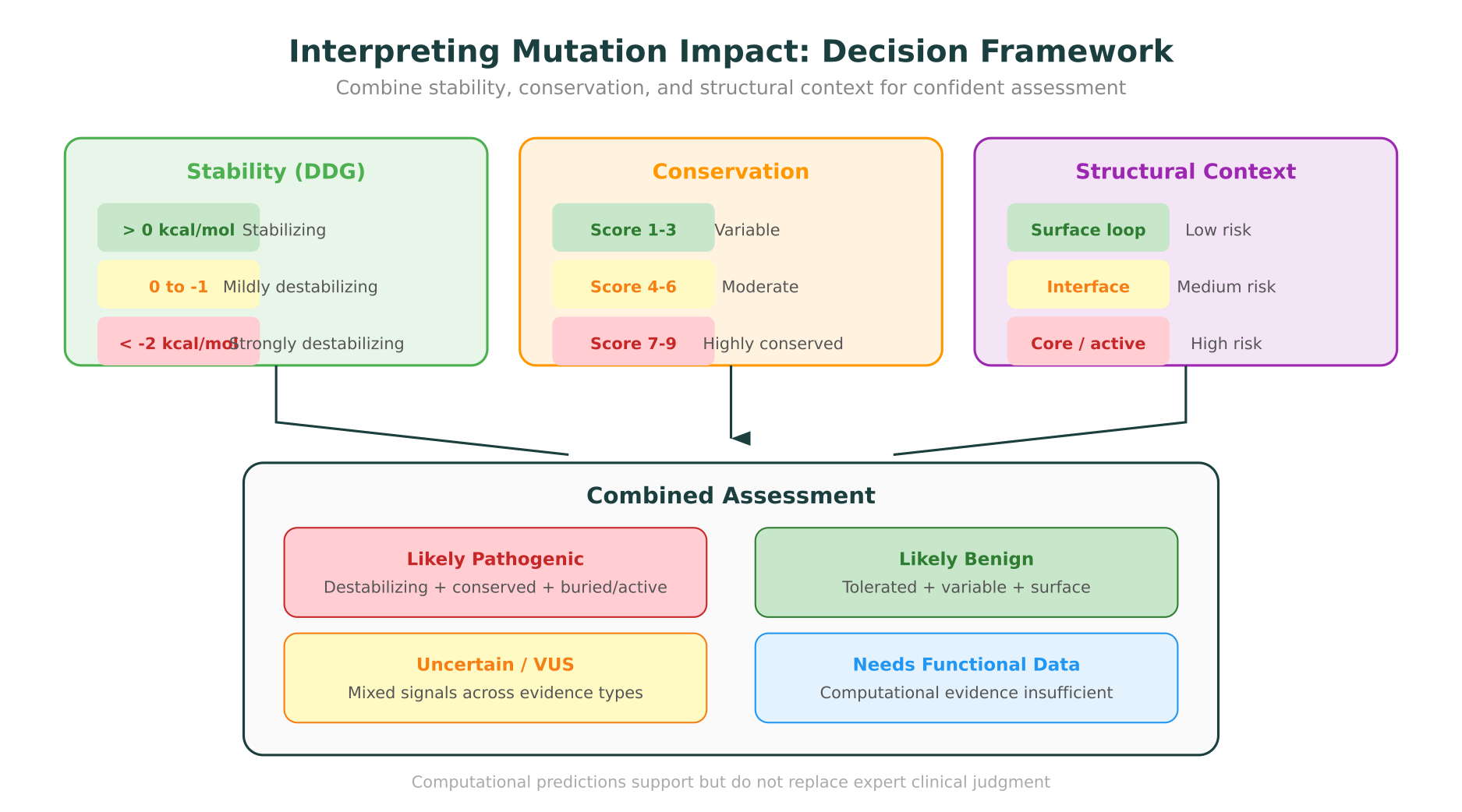
DynaMut2 predicts the change in Gibbs free energy of folding (delta-delta-G, or DDG) caused by a single-point mutation. This value tells you whether the mutation stabilizes or destabilizes the protein:
- DDG < -1 kcal/mol: The mutation is predicted to destabilize the protein. Values more negative than -2 kcal/mol are strongly destabilizing and often associated with loss of function.
- DDG near 0: The mutation has minimal effect on stability.
- DDG > 1 kcal/mol: The mutation stabilizes the protein, which can be relevant for protein engineering but may also indicate functional disruption if it locks the protein into an inactive conformation.
DynaMut2 combines Normal Mode Analysis (NMA), which models protein dynamics, with graph-based representations of protein structure. It also classifies mutations as stabilizing or destabilizing with reported accuracy in the range of 73-80% on benchmark datasets (Nucleic Acids Research, 2020).
Interpreting stability predictions
Stability predictions are most useful when combined with structural context:
- A strongly destabilizing mutation (DDG < -2 kcal/mol) in the hydrophobic core is likely pathogenic.
- A mildly destabilizing mutation on a solvent-exposed loop may be tolerated.
- A stabilizing mutation in a hinge region could impair conformational changes required for function.
Other stability prediction tools include FoldX, Rosetta ddg_monomer, and mCSM. Each uses different energy functions and structural representations. When possible, checking predictions across multiple tools increases confidence.
Step 4: Evaluate Evolutionary Conservation
Evolutionary conservation is one of the strongest predictors of functional importance. Residues conserved across millions of years of evolution are under selective pressure because they matter for protein function or structure.
ConSurf for conservation analysis
ConSurf is the standard tool for mapping conservation scores onto protein structures. It works by:
- Collecting homologous sequences through a database search (PSI-BLAST or HMMER against UniRef90)
- Building a multiple sequence alignment
- Computing position-specific conservation scores using Bayesian or maximum-likelihood methods
- Mapping scores onto the 3D structure with a color gradient (conserved = purple, variable = teal)
A highly conserved residue (ConSurf score 8-9) that is mutated to a chemically dissimilar amino acid is a strong signal for functional impact.
AlphaMissense: AI-powered conservation at scale
Google DeepMind’s AlphaMissense, published in Science in September 2023, took a different approach to conservation. Rather than aligning sequences, it fine-tuned the AlphaFold architecture to predict the pathogenicity of all 71 million possible single amino acid substitutions in the human proteome.
Key findings:
- AlphaMissense classified 89% of all possible human missense variants as either likely benign or likely pathogenic.
- It achieved an area under the ROC curve (AUROC) of 0.940 on ClinVar benchmarks, placing it among the top-performing pathogenicity predictors.
- It reclassified 32% of variants that were previously of uncertain significance.
AlphaMissense scores are now available through the AlphaFold database and provide an immediate signal for most human missense variants without running any additional analysis.
Step 5: Check Functional Domains and Known Annotations
Not all protein regions are equally important. Mutations in catalytic sites, binding interfaces, or regulatory domains carry higher prior probability of functional impact than mutations in unstructured linkers.
Key resources
- UniProt provides curated annotations for functional domains, active site residues, binding sites, signal peptides, and post-translational modification sites. The UniProt Knowledgebase (UniProtKB/Swiss-Prot) contains over 570,000 manually reviewed entries.
- InterPro classifies protein sequences into families, domains, and functional sites using predictive models from multiple databases (Pfam, PANTHER, CDD, and others). Over 47,000 entries cover the majority of known protein sequences.
- ClinVar provides a record of previously classified variants at or near the same position. If a different missense variant at the same residue has been classified as pathogenic (ACMG criterion PM5), that significantly increases the evidence for pathogenicity of a novel variant at that position.
Putting it together: a practical example
Consider a novel missense variant in BRCA1 at position Cys61Gly in the RING domain.
- Structure: The RING domain coordinates two zinc ions through conserved cysteine and histidine residues. Cys61 directly ligates one of the zinc ions. An experimental structure (PDB: 1JM7) shows the coordination geometry.
- Stability: DynaMut2 predicts strong destabilization (DDG approximately -3.5 kcal/mol) because disrupting zinc coordination collapses the domain fold.
- Conservation: ConSurf scores Cys61 at 9 (maximally conserved). The residue is invariant across all vertebrate BRCA1 orthologs.
- AlphaMissense: Classifies C61G as likely pathogenic with high confidence.
- ClinVar: Multiple other cysteine-to-non-cysteine substitutions in the RING domain are classified as pathogenic.
- Functional evidence: The RING domain is required for BRCA1’s E3 ubiquitin ligase activity. Disrupting it abolishes DNA repair function.
Each line of evidence independently supports pathogenicity. Together, they make a strong case that would satisfy multiple ACMG criteria (PS3, PM1, PM5, PP3).
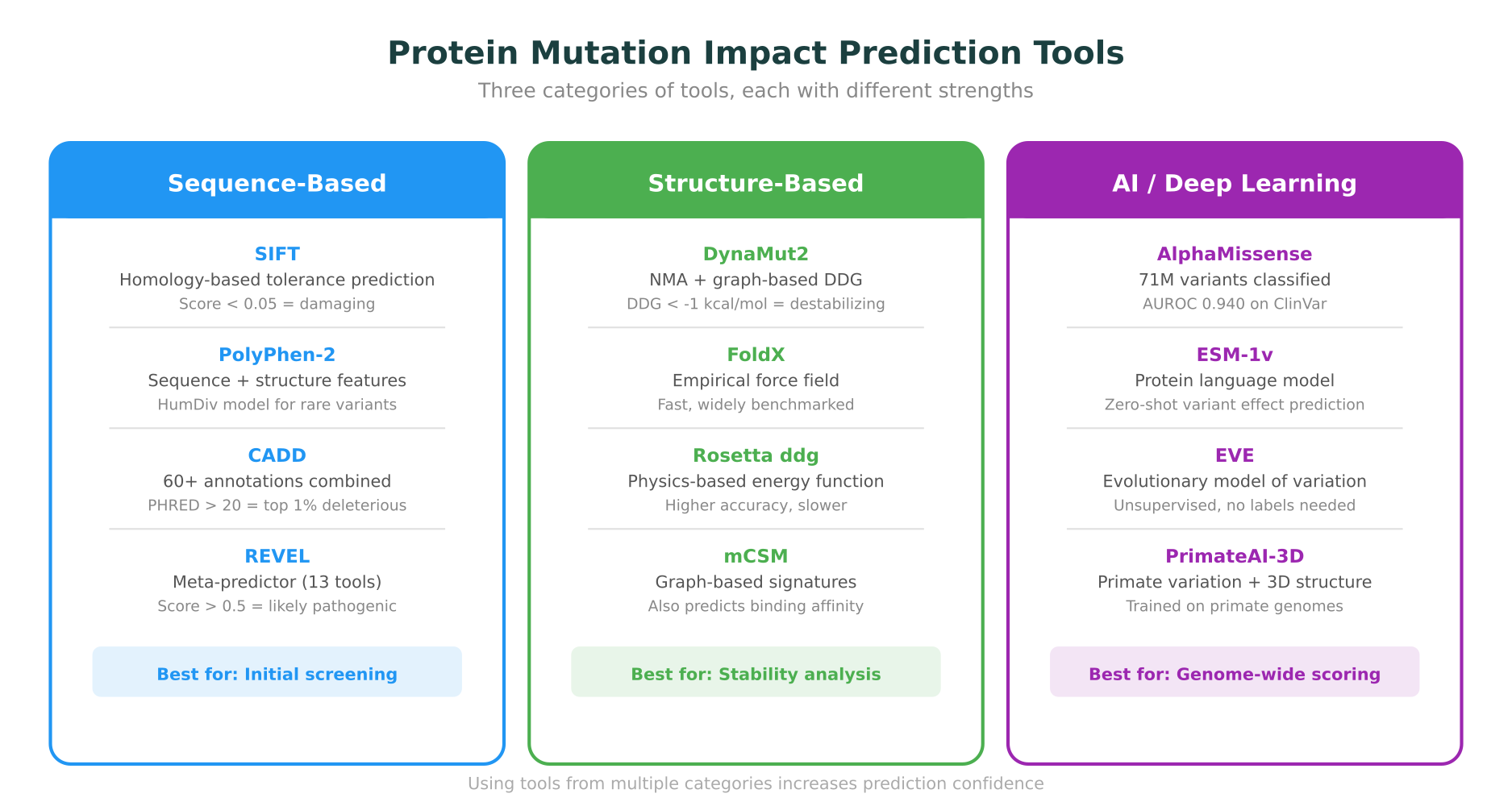
Sequence-Based Predictors: When Structure Isn’t Available
Not every analysis requires or has access to a protein structure. Several well-validated tools predict mutation impact from sequence alone.
Commonly used tools
- SIFT (Sorting Intolerant From Tolerant) predicts whether a substitution is tolerated based on sequence homology. Scores below 0.05 are predicted damaging.
- PolyPhen-2 combines sequence features with structural features (when available) to predict pathogenicity. HumDiv and HumVar are the two trained models, with HumDiv recommended for rare variants in Mendelian disease.
- CADD (Combined Annotation Dependent Depletion) integrates over 60 annotations into a single deleteriousness score. PHRED-scaled scores above 20 correspond to the top 1% most deleterious variants in the genome.
- REVEL is a meta-predictor that combines scores from 13 individual tools, and consistently ranks among the top performers in benchmarks. Scores above 0.5 suggest pathogenicity.
These tools are most useful for initial screening. When a variant scores as damaging across multiple predictors, it warrants deeper structural and functional investigation.
Bringing It All Together: Integrated Workflows
The power of protein mutation impact prediction comes from combining multiple lines of evidence. No single tool provides a definitive answer. A variant that is predicted destabilizing by DynaMut2, conserved by ConSurf, classified as likely pathogenic by AlphaMissense, and located in a known functional domain is far more confidently interpreted than one assessed by any tool in isolation.
The traditional workflow requires navigating between multiple disconnected tools: PDB or AlphaFold for the structure, a separate viewer for visualization, DynaMut2 for stability, ConSurf for conservation, UniProt for annotations, and ClinVar for prior classifications. Each tool has its own input format, and synthesizing results across them is manual and error-prone.
This is where molecular intelligence platforms fundamentally change the workflow. Purna AI’s MIP integrates structure retrieval (from PDB and AlphaFold), Molstar visualization, DynaMut2 stability analysis, conservation scoring, and queries across 30+ biological databases into a single workspace. A researcher can type a natural language query like “What is the structural impact of the p.Arg248Trp mutation in TP53?” and receive a synthesized answer that pulls structure, stability predictions, conservation data, ClinVar classifications, and functional annotations together, with citations to every source database.
This isn’t about replacing expert judgment. Variant interpretation, especially in clinical settings, requires human oversight. But the time spent gathering evidence from disconnected tools can be compressed from hours to minutes, allowing researchers and clinicians to focus on interpretation rather than data retrieval.
For teams working on protein engineering and drug discovery, the same workflow applies to evaluating designed mutations. Predicting which modifications will improve binding affinity, increase thermostability, or alter specificity requires the same structural, stability, and conservation analyses described here.
Protein mutation impact prediction is no longer a specialist-only capability. With the right tools and an integrated approach, any biology team can move from a variant call to a structural interpretation, grounded in evidence and traceable to source databases.
MIP is Purna AI’s Molecular Intelligence Platform, an AI-powered workspace for biology teams. Variant interpretation, protein structure prediction and visualization, DynaMut2 stability analysis, and 30+ database integrations in one place. Explore the platform at purna.ai. Researchers can apply for up to $10,000 in free credits to run their analyses on MIP.
Explore Purna's Molecular Intelligence Platform
AI-powered workspace for biology teams to accelerate drug discovery from target identification to lead optimization.
Try Purna AI →
