AlphaFold 3 vs Boltz-2 vs ESMFold: Protein Structure Prediction Tools Compared

Choosing the right protein structure prediction tool in 2026 is no longer a simple question. Two years after AlphaFold 2 reshaped the field, researchers now have multiple serious options: AlphaFold 3 with its expanded capabilities for biomolecular complexes, Boltz-2 as an open-source alternative with comparable accuracy, and ESMFold for rapid single-sequence inference. Each tool reflects a different set of trade-offs in accuracy, speed, licensing, and the types of biological questions it can address.
This comparison breaks down the major protein structure prediction tools available today, covering what each handles, where each excels, and practical recommendations for different research workflows. Whether you work in clinical genomics, drug discovery, or protein engineering, the right choice depends on your specific use case.
Why the Landscape Has Changed
The release of AlphaFold 2 in 2020, and its publication in Nature in 2021, set a new standard for protein structure prediction. But the field has moved well beyond single-chain monomer folding. Researchers now routinely need to predict protein-protein complexes, protein-ligand interactions, protein-nucleic acid assemblies, and the effects of post-translational modifications.
No single tool covers every scenario optimally. AlphaFold 3 expanded the scope of prediction significantly, but its licensing restrictions pushed the community to develop open alternatives. ESMFold took a fundamentally different architectural approach, trading some accuracy for dramatic speed gains. And newer entrants like Boltz-2 and Chai-1 have demonstrated that open-source models can match proprietary ones on key benchmarks.
Understanding these tools in context requires looking at the underlying architectures, what each can and cannot predict, and how they fit into real research workflows. For teams working within a molecular intelligence framework, the goal is not just running predictions but connecting them to downstream analysis: stability assessment, variant interpretation, and functional annotation.
AlphaFold 3: The Current Benchmark
AlphaFold 3, published by Google DeepMind in Nature in May 2024, introduced a fundamentally new architecture compared to its predecessor. While AlphaFold 2 used an Evoformer-based approach with structure modules that iteratively refined coordinates, AlphaFold 3 adopted a diffusion-based generative model for the final structure generation step.
Architecture and approach
The key architectural shift in AlphaFold 3 is the replacement of the structure module with a diffusion network. Instead of directly predicting atomic coordinates through iterative refinement, AlphaFold 3 generates structures by denoising from random noise, similar to how image diffusion models work. This approach naturally handles the uncertainty inherent in flexible regions and allows the model to generate diverse conformations.
AlphaFold 3 retains an updated version of the Evoformer trunk for processing multiple sequence alignments (MSAs) and template information. The MSA processing stage extracts evolutionary covariance signals that inform the diffusion model about likely inter-residue contacts.
What AlphaFold 3 can predict
The most significant expansion in AlphaFold 3 is its ability to model biomolecular complexes beyond just proteins:
- Protein-protein complexes: Multimer prediction with improved accuracy over AlphaFold-Multimer
- Protein-ligand interactions: Small molecule binding poses, though accuracy varies with ligand complexity
- Protein-nucleic acid complexes: DNA and RNA binding, relevant for transcription factor studies
- Ion and cofactor placement: Metal ions and small cofactors within binding sites
- Post-translational modifications: Glycosylation, phosphorylation, and other covalent modifications
On the CASP15 benchmarks, AlphaFold 3 demonstrated improved performance on protein complexes compared to AlphaFold-Multimer, and was the first tool to show competitive accuracy on protein-ligand and protein-nucleic acid targets in a blind assessment setting.
Limitations and licensing
AlphaFold 3’s licensing has been one of its most discussed constraints. Google DeepMind released the model weights under an academic-only license, and the AlphaFold Server web interface imposes daily job limits and restricts commercial use. For pharmaceutical companies and biotech startups, this creates a practical barrier.
Additionally, while AlphaFold 3 handles a broad range of biomolecular complexes, its ligand docking accuracy does not consistently match specialized docking tools like Glide or GOLD for challenging drug-like molecules. The protein-ligand predictions are useful for understanding binding modes but may not be precise enough for lead optimization workflows.
The MSA generation step also adds computational overhead. For a typical prediction, the MSA search against sequence databases (UniRef, BFD, MGnify) can take minutes to hours depending on the target, before the diffusion model even begins generating the structure.
Boltz-2: The Open-Source Challenger
Boltz-2, developed by the Boltz team and released under the MIT license, emerged as a direct response to the accessibility limitations of AlphaFold 3. It is a fully open-source biomolecular structure prediction model that matches or approaches AlphaFold 3 accuracy across multiple benchmarks while removing all commercial use restrictions.
Architecture and approach
Boltz-2 builds on a similar diffusion-based architecture to AlphaFold 3 but with several engineering improvements. The model uses a modified transformer trunk for processing input features and a diffusion module for structure generation. Notably, Boltz-2 made MSA computation optional rather than required, allowing users to trade some accuracy for significantly faster predictions when evolutionary information is not critical.
The model was trained on a large dataset of experimentally determined structures from the PDB, supplemented with distillation from predicted structures. This training approach allows Boltz-2 to generalize across protein families even when experimental coverage is sparse.
Key capabilities
Boltz-2 supports a comparable feature set to AlphaFold 3:
- Protein monomers and complexes: Full support for multimer prediction
- Protein-ligand complexes: Small molecule binding with support for diverse ligand chemistry
- Covalent modifications: Post-translational modifications and covalent ligand attachments
- Nucleic acid complexes: DNA and RNA binding predictions
- Confidence estimation: Per-residue and per-atom confidence scores for assessing prediction reliability
One of Boltz-2’s distinguishing features is its support for covalent modifications in a more flexible manner than AlphaFold 3. Researchers working with covalent inhibitors or heavily modified proteins have reported better handling of these edge cases.
Why licensing matters
The MIT license is a significant differentiator. For academic researchers, the practical difference may seem small, since AlphaFold 3 is available for non-commercial use. But for anyone working at a pharmaceutical company, a biotech startup, or a CRO, the ability to run Boltz-2 on internal infrastructure without licensing concerns is a meaningful advantage.
This also matters for reproducibility and customization. With full access to the source code and weights, researchers can fine-tune Boltz-2 on proprietary datasets, modify the architecture for specific applications, or integrate it into automated pipelines without legal review.
ESMFold: Speed Through Simplicity
ESMFold, developed by Meta AI and published in Science in 2023, took a fundamentally different approach to the structure prediction problem. Instead of relying on MSAs and coevolutionary signals, ESMFold uses a protein language model (ESM-2) to extract structural information directly from a single input sequence.
Architecture and approach
ESMFold’s architecture consists of two main components: the ESM-2 protein language model and a folding module that converts language model representations into 3D coordinates. ESM-2 was trained on millions of protein sequences using masked language modeling, a self-supervised objective that forces the model to learn the statistical patterns of protein sequences, including implicit structural information.
The critical insight behind ESMFold is that a sufficiently large language model, trained on enough protein sequences, learns to encode structural information without ever seeing a 3D structure during pretraining. The folding module then translates these learned representations into atomic coordinates using an architecture inspired by AlphaFold 2’s structure module.
The speed advantage
The absence of MSA computation is ESMFold’s most important practical feature. For a typical protein, MSA generation involves searching against databases containing hundreds of millions of sequences. This search can take anywhere from minutes to hours depending on the protein length, the database size, and the available hardware.
ESMFold skips this entirely. Given a single amino acid sequence, it produces a structure prediction in seconds on a modern GPU. This makes it practical for use cases that are simply infeasible with MSA-dependent methods:
- Metagenomic screening: Predicting structures for millions of sequences from environmental samples
- Protein engineering campaigns: Rapidly assessing thousands of designed variants
- Real-time analysis: Embedding structure prediction into interactive research workflows
- Large-scale proteome annotation: Structural characterization of entire proteomes
Accuracy trade-offs
The trade-off for this speed is accuracy. On CASP15 targets, ESMFold’s performance was below AlphaFold 3 and Boltz-2, particularly on targets with few homologs in sequence databases. For proteins from well-studied families with abundant evolutionary information, the accuracy gap narrows. For orphan proteins or targets requiring precise side-chain placement, the gap is more substantial.
ESMFold is best understood as a rapid screening tool rather than a precision prediction engine. Its pLDDT confidence scores are well-calibrated, meaning researchers can use them to identify which predictions are likely reliable and which should be refined with a more accurate (and slower) method.
Limitations
Beyond accuracy, ESMFold has hard constraints on what it can predict:
- No complex prediction: ESMFold is limited to single-chain proteins. It cannot predict protein-protein interfaces, protein-ligand binding, or protein-nucleic acid complexes.
- No ligand or cofactor support: The model has no mechanism for handling non-protein entities.
- Sequence length limits: Performance degrades for very long sequences (typically above 1,000 residues).
For workflows involving protein mutation impact prediction, ESMFold can provide a rapid initial structure, but the analysis of mutation effects at interfaces or binding sites requires tools that handle complexes.
Head-to-Head Comparison
The following table summarizes the key differences across the three primary tools.
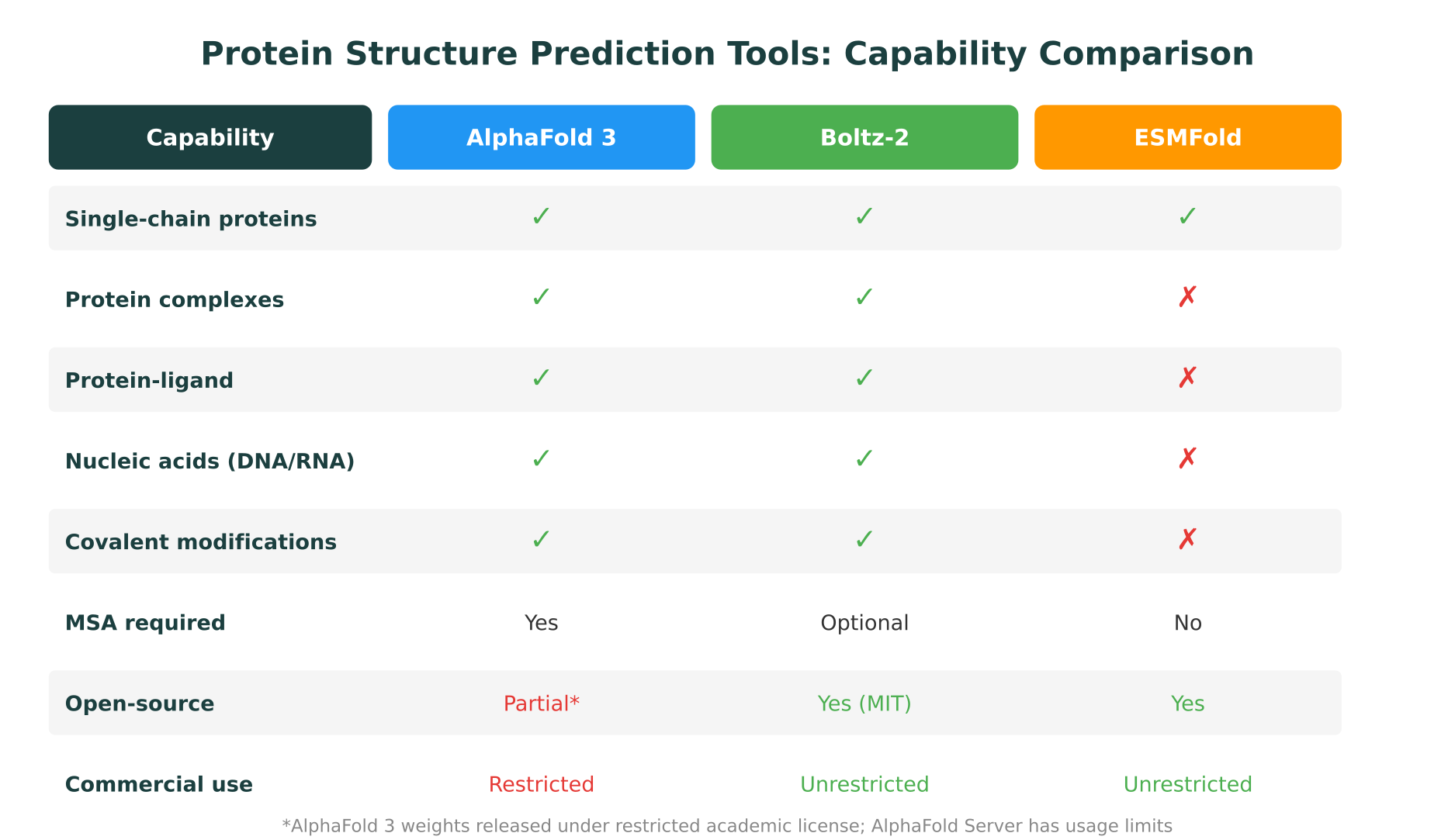
| Feature | AlphaFold 3 | Boltz-2 | ESMFold |
|---|---|---|---|
| Developer | Google DeepMind | Boltz (community) | Meta AI |
| Architecture | Evoformer + diffusion | Transformer + diffusion | ESM-2 pLM + folding module |
| MSA required | Yes | Optional | No |
| Single-chain accuracy | Highest | Comparable | Moderate |
| Complex prediction | Yes | Yes | No |
| Protein-ligand | Yes | Yes | No |
| Nucleic acids | Yes | Yes | No |
| Covalent mods | Yes | Yes (flexible) | No |
| Inference speed | Minutes to hours | Minutes | Seconds |
| Open-source | Partial (restricted weights) | Yes (MIT) | Yes |
| Commercial use | Restricted | Unrestricted | Unrestricted |
| Key publication | Nature, 2024 | Preprint, 2025 | Science, 2023 |
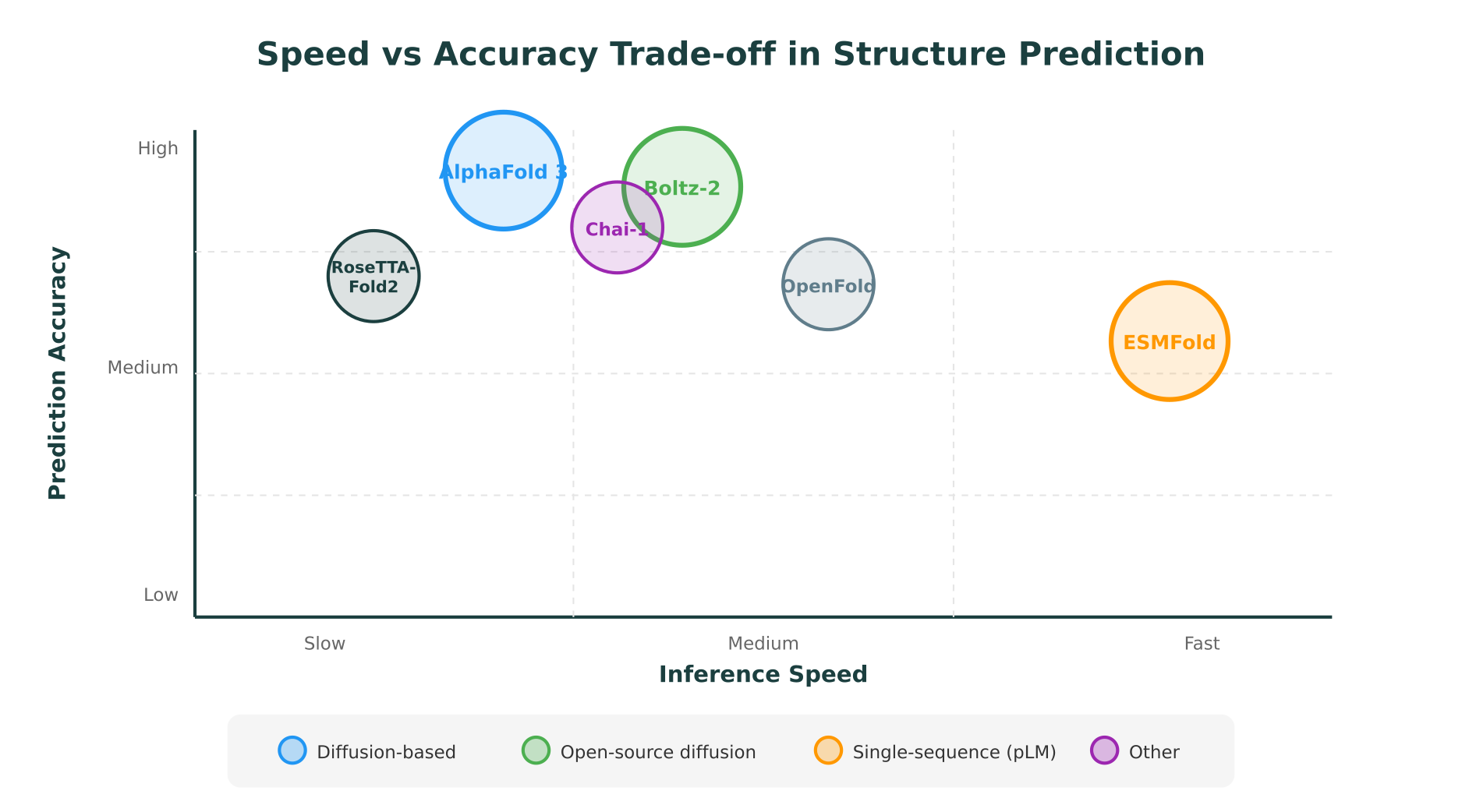
Accuracy benchmarks
Direct accuracy comparisons require careful context. On CASP15 monomer targets, all three tools produce high-quality predictions for proteins with abundant homologs (GDT-TS scores above 80 for most targets). The differences emerge on harder cases:
- Orphan proteins (few homologs): AlphaFold 3 and Boltz-2 significantly outperform ESMFold, since MSA-derived coevolutionary signals remain the strongest predictor of contact maps for distant homology cases.
- Multimeric complexes: Only AlphaFold 3 and Boltz-2 can make these predictions. AlphaFold 3 currently leads on interface accuracy (DockQ scores), with Boltz-2 close behind.
- Protein-ligand binding: Both AlphaFold 3 and Boltz-2 can predict ligand binding poses, though neither consistently matches physics-based docking tools for drug-like molecules. Boltz-2 has shown slightly better handling of covalent ligands in community benchmarks.
Practical throughput
For a 300-residue single-chain protein on a single A100 GPU:
- ESMFold: Approximately 5-10 seconds (no MSA step)
- Boltz-2 (without MSA): Approximately 1-3 minutes
- Boltz-2 (with MSA): Approximately 10-30 minutes (including MSA generation)
- AlphaFold 3: Approximately 15-45 minutes (including MSA generation)
These timings scale roughly linearly with sequence length for ESMFold, and quadratically for the MSA-dependent methods due to the attention mechanisms over aligned sequences.
Other Tools Worth Knowing
While AlphaFold 3, Boltz-2, and ESMFold represent the three main paradigms, several other tools fill important niches.
RoseTTAFold2
Developed by David Baker’s group at the University of Washington (published in Science, 2023), RoseTTAFold2 uses a three-track neural network architecture that simultaneously processes sequence, pairwise distance, and 3D coordinate information. It supports protein complexes and has been particularly valued in the protein design community, where it integrates with RFdiffusion for de novo protein design workflows.
RoseTTAFold2’s accuracy falls between AlphaFold 3 and ESMFold on most benchmarks. Its main advantage is tight integration with the Rosetta suite of protein design and analysis tools.
OpenFold
OpenFold is a faithful open-source reimplementation of AlphaFold 2, maintained by a consortium of academic labs and industry partners. It does not replicate AlphaFold 3’s diffusion-based architecture or complex prediction capabilities, but it provides a well-documented, trainable codebase for researchers who want to understand or modify the AlphaFold 2 architecture.
OpenFold has been particularly useful for training custom models on specialized protein families and for educational purposes. Its accuracy matches AlphaFold 2 closely, which means it remains competitive for single-chain monomer prediction.
Chai-1
Chai-1, developed by Chai Discovery, is a more recent entry that supports biomolecular complex prediction with an architecture influenced by AlphaFold 3. It handles proteins, nucleic acids, small molecules, and covalent modifications. Early benchmarks suggest accuracy comparable to Boltz-2, with the added feature of built-in confidence calibration and a commercial API.
Chai-1 is particularly notable for its focus on drug discovery applications, with specific optimizations for protein-ligand binding accuracy. It is available under a more permissive license than AlphaFold 3, though not as open as Boltz-2’s MIT license.
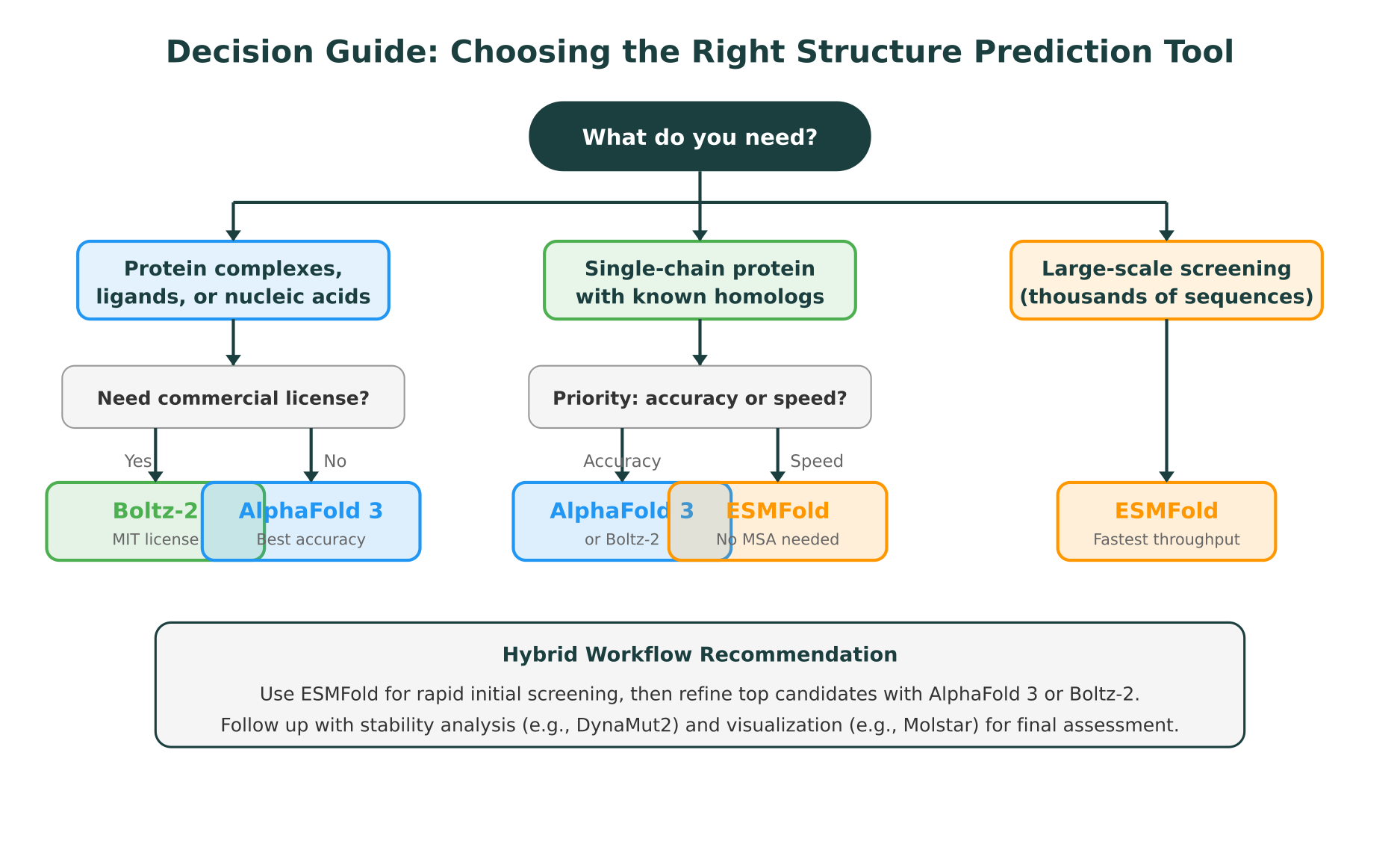
Practical Recommendations: When to Use Each Tool
The best tool depends on what you are trying to accomplish. Here are concrete recommendations for common research scenarios.
Use AlphaFold 3 when:
- You need the highest possible accuracy for a single high-value target
- You are predicting protein-nucleic acid complexes
- You are in an academic setting with no commercial use requirements
- You need the credibility of the most widely validated tool for publications
Use Boltz-2 when:
- You need to predict complexes with ligands, nucleic acids, or modifications
- You need commercial freedom (pharma, biotech, CRO environments)
- You want to fine-tune or customize the model for specific protein families
- You want to integrate structure prediction into automated pipelines
- You need MSA-free mode for faster turnaround without sacrificing complex prediction
Use ESMFold when:
- You are screening thousands or millions of sequences and need rapid throughput
- You are working with metagenomic data or protein engineering libraries
- You only need single-chain structure predictions
- You are building real-time or interactive analysis tools
- You need a quick initial structure before running detailed downstream analysis
Hybrid workflows
In practice, many research groups use a combination of tools. A common pattern is to run ESMFold first for rapid screening across a large set of candidates, filter based on pLDDT confidence scores, and then run AlphaFold 3 or Boltz-2 on the top candidates for higher-accuracy predictions. This two-stage approach balances throughput with accuracy.
For drug discovery workflows, the combination of Boltz-2 for initial complex prediction followed by physics-based refinement (molecular dynamics, free energy perturbation) is becoming a standard practice. The AI-predicted structure provides a starting point, and physics-based methods refine the binding pose and estimate binding affinity.
Licensing Comparison for Commercial Teams
Licensing is one of the most consequential practical differences between these tools. The following breakdown matters for any team operating outside of purely academic research.
| Tool | License | Commercial Use | Self-hosting | Fine-tuning |
|---|---|---|---|---|
| AlphaFold 3 | Academic-only (weights), CC-BY-NC (server) | No | Limited | No |
| Boltz-2 | MIT | Yes | Yes | Yes |
| ESMFold | MIT | Yes | Yes | Yes |
| RoseTTAFold2 | BSD | Yes | Yes | Yes |
| OpenFold | Apache 2.0 | Yes | Yes | Yes |
| Chai-1 | Proprietary + API | Via API | No (API only) | No |
For pharmaceutical companies evaluating these tools, Boltz-2’s MIT license is the most permissive option that still offers AlphaFold 3-class capabilities (complex prediction, ligand binding, nucleic acids). ESMFold is equally permissive but limited to single-chain predictions.
Connecting Structure Prediction to Downstream Analysis
Predicting a structure is rarely the end of a research workflow. The predicted model typically feeds into stability analysis, mutation impact assessment, molecular docking, or functional annotation. How well a prediction tool integrates with these downstream steps matters as much as raw accuracy.
For variant interpretation, a predicted structure enables assessment of whether a mutation disrupts the hydrophobic core, interferes with a binding interface, or destabilizes secondary structure elements. Tools like DynaMut2 quantify the predicted change in thermodynamic stability (delta-delta-G) upon mutation, and conservation analysis from multiple sequence alignments adds evolutionary context. As covered in our guide on protein mutation impact prediction, combining structural, stability, and conservation evidence produces more reliable assessments than any single line of evidence alone.
For teams working across multiple biological data types, from genomics to proteomics to transcriptomics, structure prediction is one component of a broader analytical picture. A variant identified through exome sequencing may need structural context from AlphaFold, stability analysis from DynaMut2, population frequency data from gnomAD, and clinical significance from ClinVar, all synthesized into a coherent interpretation.
Purna AI’s Molecular Intelligence Platform (MIP) integrates this kind of multi-step analysis into a single workspace. MIP pulls structures from the PDB and the AlphaFold database, renders them in Molstar for interactive visualization, and runs DynaMut2 for stability analysis. Rather than switching between separate tools for each step, researchers can move from structure retrieval to mutation analysis to variant classification in one environment. The platform supports querying over 30 clinical and biological databases through natural language, so the connection between a structural prediction and its clinical or functional context is immediate.
This is the broader shift that molecular intelligence represents: not replacing any single prediction tool, but providing the connective layer that makes each tool’s output more useful by linking it to everything else a researcher needs to make a decision.
What to Watch in 2026 and Beyond
The protein structure prediction landscape continues to evolve rapidly. Several trends are worth tracking.
Diffusion models as the default. The shift from iterative refinement (AlphaFold 2) to diffusion-based generation (AlphaFold 3, Boltz-2) appears to be permanent. Diffusion models handle uncertainty more naturally and extend more cleanly to complex prediction. Expect future tools to build on this paradigm.
MSA-free methods improving. ESMFold demonstrated that protein language models encode meaningful structural information. As language models scale and training data grows, MSA-free methods will continue to close the accuracy gap with MSA-dependent approaches. This is particularly important for metagenomic and protein engineering applications where speed is critical.
Confidence calibration becoming standard. All major tools now provide confidence scores (pLDDT, pAE, pDockQ), and researchers are increasingly using these to filter predictions before downstream analysis. Tools with better-calibrated confidence scores will have an edge, because knowing when a prediction is unreliable is as valuable as the prediction itself.
Integration over isolation. The trend is toward platforms that connect structure prediction with stability analysis, variant interpretation, and functional annotation. Standalone prediction tools will remain important for benchmarking and methods development, but for applied research, integrated workflows reduce friction and errors.
For an overview of how AI is being applied across biology beyond just structure prediction, including genomics, drug discovery, and clinical interpretation, see our broader analysis.
Conclusion
AlphaFold 3, Boltz-2, and ESMFold represent three distinct approaches to protein structure prediction, each optimized for different trade-offs. AlphaFold 3 remains the accuracy leader for biomolecular complexes. Boltz-2 matches that capability with full open-source access and commercial freedom. ESMFold offers unmatched speed for single-chain predictions at the cost of accuracy and scope.
The right choice is not about which tool is “best” in the abstract. It depends on whether you need complex prediction or single-chain only, whether speed or accuracy is the priority, whether you need commercial licensing, and how the prediction fits into your broader analytical workflow.
For researchers who want to connect structure prediction to variant interpretation, stability analysis, and clinical database lookups in one workspace, Purna AI’s MIP brings these capabilities together. Academic researchers can apply for up to $10,000 in free MIP credits to explore these workflows on their own data.
Purna AI’s Molecular Intelligence Platform (MIP) is an AI-powered workspace for biology teams. It brings together protein structure prediction, variant interpretation, stability analysis, and 30+ clinical database integrations into one environment. Built for teams who work with structural and genomic data and need consistent, reproducible answers without juggling disconnected tools. Learn more at purna.ai.
Explore Purna's Molecular Intelligence Platform
AI-powered workspace for biology teams to accelerate drug discovery from target identification to lead optimization.
Try Purna AI →
