Bioinformatics Without Coding: How AI Is Making Computational Biology Accessible

Bioinformatics has a gatekeeping problem. The field sits at the intersection of biology and computer science, but most of the people who need its outputs are biologists, not programmers. A wet lab scientist studying gene expression, a clinical geneticist interpreting variants, a biochemist analyzing protein interactions: all of them depend on computational analysis, and most of them cannot write the code to run it.
This is not a new observation. The gap between biological expertise and computational capability has been documented for over a decade. What is new is that practical solutions are starting to emerge. Natural language interfaces, AI-powered analysis platforms, and integrated workspaces are making bioinformatics without coding a genuine possibility, not by dumbing down the science, but by changing how researchers interact with computational tools.
The Scale of the Accessibility Problem
The numbers tell a clear story. A 2023 survey published in PLOS Computational Biology found that over 60% of life science researchers reported needing bioinformatics analysis for their work, but fewer than 25% felt confident performing it independently. The rest relied on collaborators, core facilities, or simply did not run analyses they knew would be valuable.
This gap has consequences. Research projects stall while waiting for bioinformatics support. Hypotheses go untested because the analysis would require scripting skills the researcher does not have. Clinical labs struggle to scale variant interpretation because each case requires manual expert review.
The problem is not intelligence or motivation. A molecular biologist who can design a CRISPR experiment, culture cells, and interpret Western blots is not incapable of understanding computational analysis. The barrier is the interface: command-line tools, programming languages, package management, environment configuration, and the debugging that comes with all of it.
What “bioinformatics” actually requires today
Consider what a typical bioinformatics task involves. A researcher wants to understand whether a set of variants identified in a patient’s exome are clinically significant. To do this properly, they need to:
- Query ClinVar for existing classifications
- Check population frequencies in gnomAD
- Look up gene-disease associations in OMIM
- Assess protein impact using tools like SIFT, PolyPhen, or CADD
- Examine protein structure for variants near functional domains
- Synthesize all of this into an ACMG/AMP classification
In a traditional workflow, each of these steps requires either a separate web interface with its own query syntax, or a script that calls the relevant API. A researcher fluent in Python or R can automate this. Everyone else faces hours of manual database lookups, copy-pasting between browser tabs, and the risk of missing a critical data source.
This pattern repeats across virtually every area of computational biology: genomics, proteomics, and transcriptomics all involve multi-step analyses that traditionally require scripting.
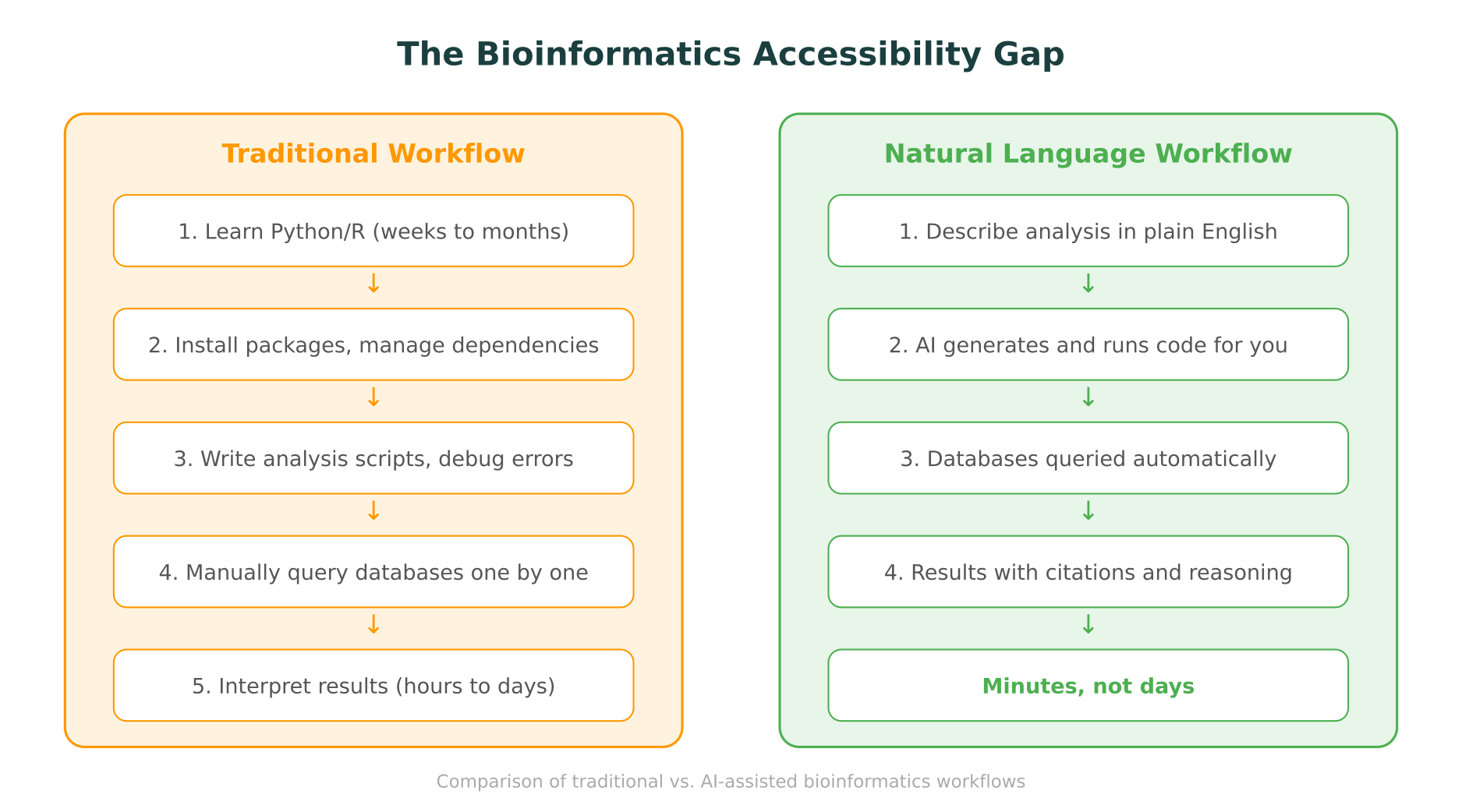
Why Existing Platforms Haven’t Solved This
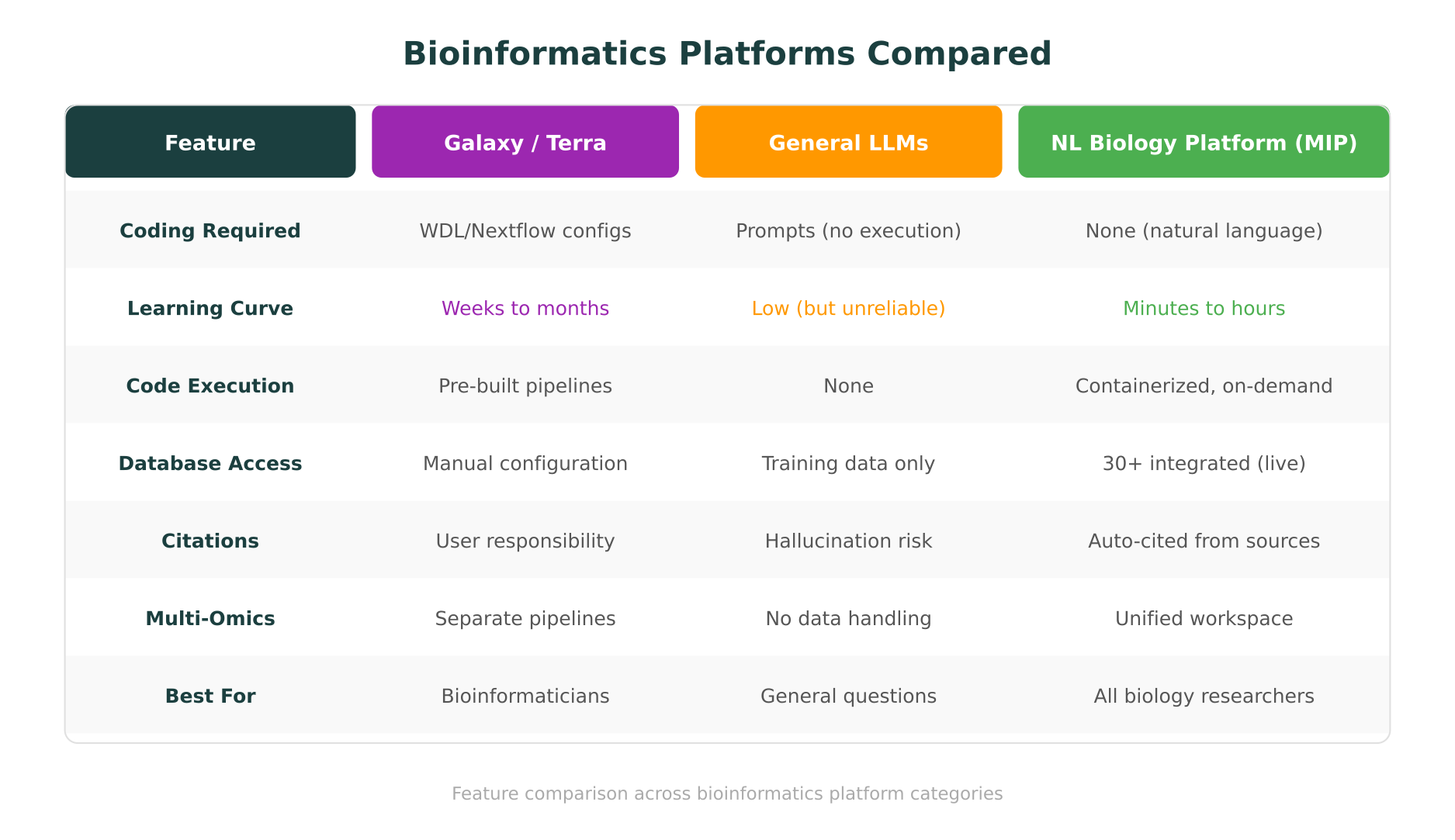
Several platforms have tried to make bioinformatics more accessible. Galaxy, Terra, and similar workflow engines deserve credit for moving computational biology forward. But they have not closed the accessibility gap, and understanding why is important.
Galaxy: powerful but complex
Galaxy, first published in Genome Research in 2005 and continuously developed since, provides a web-based interface for running bioinformatics workflows. It has a large library of tools, supports workflow sharing, and has an active community. For bioinformaticians who want to make their pipelines reproducible and shareable, it is genuinely useful.
But Galaxy is not “no-code” in the way most biologists need. Setting up a workflow still requires understanding the tools you are chaining together, configuring parameters, managing input formats, and troubleshooting when steps fail. The learning curve, while lower than pure command-line work, is still measured in weeks for most wet lab researchers. A 2022 user study from the Galaxy community itself acknowledged that new users frequently struggle with data formatting and tool selection.
Terra: cloud-native, cloud-complex
Terra, built by the Broad Institute and Verily, takes a different approach. It provides cloud-based infrastructure for running genomic analyses at scale, integrating with Google Cloud Platform and supporting WDL (Workflow Description Language) pipelines.
For large-scale projects, institutions running thousands of genomes, or multi-site collaborations, Terra provides real infrastructure value. But it assumes a level of computational literacy that most bench scientists do not have. Writing WDL workflows, configuring cloud compute resources, and managing data in cloud buckets are not tasks a researcher can pick up in an afternoon.
General-purpose LLMs: helpful but insufficient
The rise of large language models like ChatGPT and Claude has given researchers a new option: ask an AI to write the code for you. This works, sometimes. A well-prompted LLM can generate a Python script to parse a VCF file or query a database API.
The problems are well-documented. General-purpose LLMs are not connected to biological databases. They cannot execute code. They hallucinate database entries, gene names, and variant classifications. They have no way to verify their outputs against current data. A script that looks correct but queries an outdated API endpoint, or a variant classification that sounds authoritative but has no basis in ClinVar, can be worse than no answer at all.
For bioinformatics, the issue is not that LLMs are bad at code generation. It is that code generation alone is not enough. The value is in the execution, the data access, and the domain-specific reasoning that connects them.
Purna’s Molecular Intelligence Platform: no-code bioinformatics in practice
Purna AI’s Molecular Intelligence Platform (MIP) takes a fundamentally different approach from both workflow engines and general-purpose LLMs. Where Galaxy asks you to assemble pipelines and ChatGPT asks you to run the code it generates, MIP handles the entire loop: you describe what you need in plain language, and the platform reasons through the problem, queries the relevant databases, executes code in containerized environments, and returns cited results. No pipeline configuration. No copy-pasting scripts. No switching between browser tabs.
What makes this work is the depth of integration. MIP connects to over 30 clinical and biological databases, including ClinVar, gnomAD, OMIM, UniProt, PDB, PharmGKB, and LOVD, and queries them live. When you ask about a variant, the platform does not retrieve a cached answer from training data. It pulls current entries from authoritative sources and traces every conclusion back to a specific database record.
The platform also handles tasks that general LLMs simply cannot. Ask MIP to assess the structural impact of a protein mutation, and it retrieves the 3D structure from PDB or AlphaFold, renders it in Molstar for interactive visualization, runs DynaMut2 for stability analysis (reporting delta-delta-G values), and checks functional domain annotations, all from a single natural language query. Ask it to classify a variant, and it applies the full ACMG/AMP framework with structured reasoning and evidence for each criterion. These are not summaries generated from training text. They are analyses executed in real time against current data.
For researchers who do have coding skills, MIP does not impose a ceiling. The containerized code execution environment supports Python, R, and standard bioinformatics packages. You can write and run custom scripts directly in the platform, or inspect and modify the code that MIP generates. This is what makes it a genuine molecular intelligence workspace rather than a simplified chatbot: it scales from “I have never written a line of code” to “I want to run a custom differential expression pipeline,” without switching tools.
If Galaxy is a workbench that requires assembly instructions, and general LLMs are a knowledgeable friend who cannot actually do the experiment, MIP is the fully equipped lab where you describe what you need and the work gets done, documented and reproducible, while you focus on the science.
How Natural Language Interfaces Change the Workflow
The shift happening now is not about removing code from bioinformatics. It is about changing who writes it and how it gets executed.
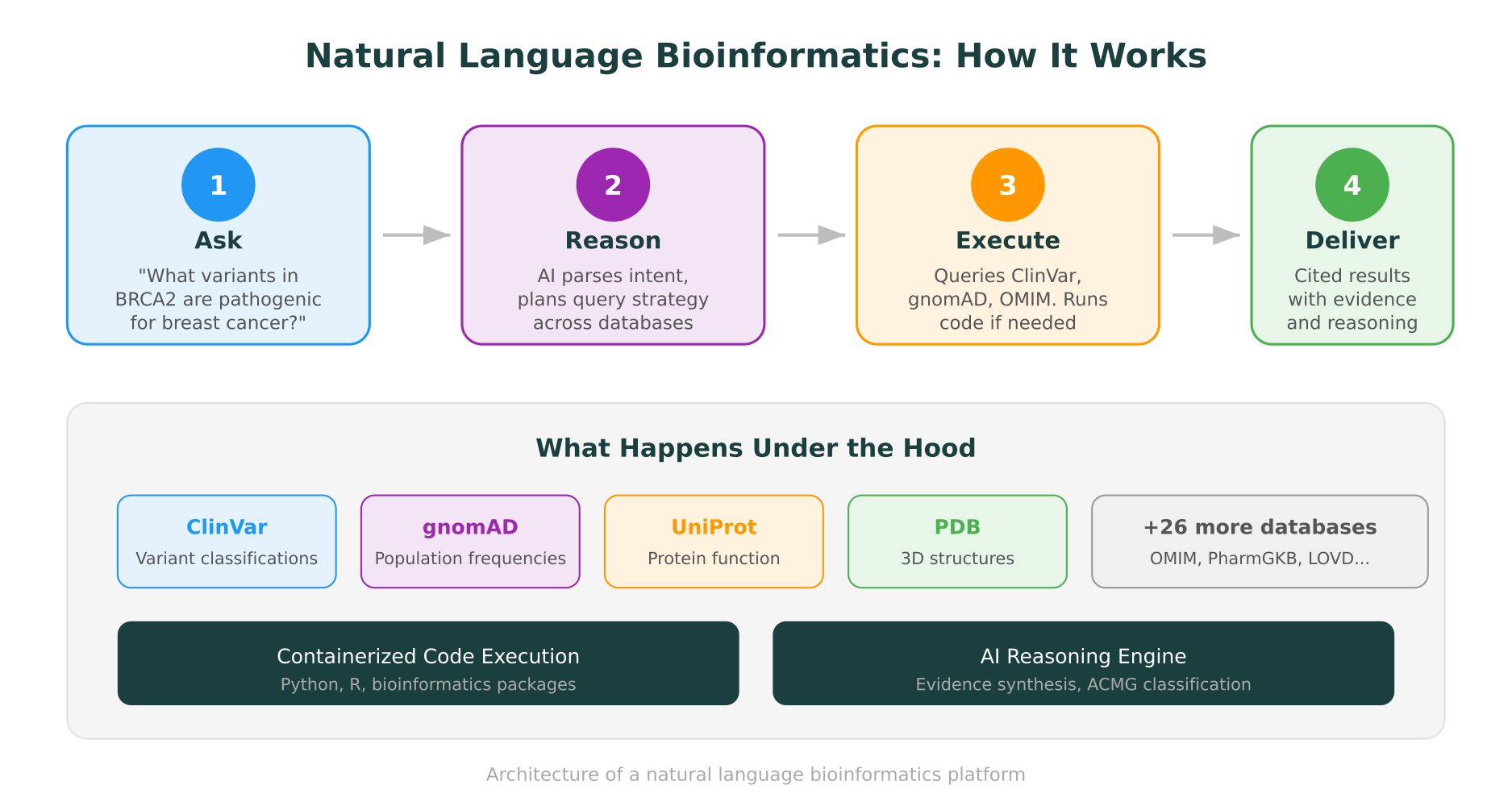
A natural language bioinformatics interface works differently from a chatbot that generates scripts. Instead of producing code for the user to run elsewhere, it takes a plain-language query, determines what databases need to be consulted, what analyses need to run, and what code (if any) needs to execute, then does all of it within a controlled environment and returns results with citations.
This is a meaningful architectural difference. The user describes what they want to know. The system handles how to get there.
What this looks like in practice
A clinical geneticist might type: “Classify the variant NM_000059.4:c.5946delT in BRCA2 using ACMG criteria.”
In a natural language bioinformatics platform, this single request triggers a cascade of operations:
- The system identifies the variant and maps it to genomic coordinates
- It queries ClinVar for existing classifications and review status
- It checks gnomAD for population allele frequency
- It retrieves functional impact predictions from multiple algorithms
- It pulls the protein structure to assess proximity to functional domains
- It synthesizes the evidence into ACMG/AMP criteria with explicit reasoning for each criterion applied
- It returns the classification with citations to every data source consulted
The geneticist did not write a single line of code. But the analysis is rigorous, traceable, and grounded in current database content, not in training data from two years ago.
What “No-Code Bioinformatics” Actually Means
The phrase “no-code” can be misleading. In software development, no-code platforms let users build applications through visual interfaces instead of programming. In bioinformatics, the concept is different.
No-code bioinformatics does not mean that no code runs. It means the researcher does not need to write, debug, or maintain the code. The computational analysis still happens, often involving the same tools and algorithms that a bioinformatician would use. The difference is in who orchestrates it.
This distinction matters because it addresses a common objection: that removing coding from bioinformatics reduces rigor. The opposite can be true. When an AI system runs a standardized analysis pipeline, applies consistent parameters, and documents every step with citations, the result can be more reproducible than a manually written script that varies between analysts.
The code execution layer
A robust no-code bioinformatics platform still needs the ability to execute code. Some analyses are inherently computational: running a multiple sequence alignment, calculating conservation scores, generating a phylogenetic tree, or performing differential expression analysis on RNA-seq data.
The key is containerized execution. The platform generates the code, runs it in an isolated environment with the necessary packages pre-installed, and returns the results. The researcher can inspect the code if they want to, but they do not need to. They can also modify it if they have the skills, which means the platform does not create a ceiling for more technical users.
This is similar to how an IDE for software engineering works. A developer using an IDE still writes code, but the IDE handles compilation, dependency resolution, debugging tools, and deployment. A molecular intelligence platform does the same for biology: it handles the computational infrastructure so the researcher can focus on the science.
Real-World Scenarios: Bioinformatics for Non-Programmers
To make this concrete, consider several scenarios where no-code bioinformatics changes the workflow for researchers who cannot (or should not need to) write code.
Scenario 1: Variant interpretation at scale
A clinical genomics lab processes 50 exomes per week. Each exome produces hundreds of variants that need classification. The traditional approach involves a bioinformatician running filtering scripts, then a clinical geneticist manually reviewing the top candidates against multiple databases.
With a natural language interface, the geneticist can query variants directly: “Show me all rare, protein-altering variants in cardiac arrhythmia genes for this patient.” The system filters the VCF, cross-references with disease gene panels, checks population frequencies, and presents the results with preliminary ACMG classifications. The geneticist reviews the evidence and makes the final call.
The bioinformatician is not replaced. They are freed to work on pipeline development and custom analyses instead of running routine queries.
Scenario 2: Protein mutation analysis
A structural biologist identifies a mutation in a protein they are studying and wants to understand its likely impact. In a coding-dependent workflow, they would need to download the PDB structure, run stability prediction software, check conservation databases, and cross-reference with known disease associations.
With an AI-powered platform, they type: “What is the structural impact of the p.Arg248Trp mutation in TP53?” The system retrieves the crystal structure, runs stability predictions using tools like DynaMut2, checks the mutation’s position relative to the DNA-binding domain, looks up clinical significance in ClinVar, and reports that this is a well-characterized hotspot mutation with destabilizing effects on protein-DNA interaction.
Scenario 3: Multi-omics exploration
A PhD student working on a rare metabolic disorder has RNA-seq data showing differential expression in several genes, plus whole-exome sequencing data from the same patient. They want to look for variants in the differentially expressed genes that might explain the expression changes.
Traditionally, this requires writing scripts to cross-reference two different data types, annotate variants, and check for regulatory effects. With a natural language interface, the student can describe the analysis: “Find coding variants in the top 20 differentially expressed genes and assess whether any could affect splicing or protein function.” The system handles the integration across multiple omics layers, applying appropriate tools at each step.
What Makes a Good No-Code Bioinformatics Platform
Not all approaches to accessible bioinformatics are equally effective. Based on where previous tools have fallen short, a few characteristics distinguish platforms that genuinely close the accessibility gap.
Live database connections
Training data goes stale. ClinVar is updated weekly. gnomAD releases new population data. OMIM adds new gene-disease associations. A platform that reasons from a static snapshot of biological knowledge will produce outdated answers. Live connections to authoritative databases, with results cited to specific entries and versions, are not optional.
Domain-specific reasoning
General-purpose AI systems do not understand ACMG criteria, protein domain architecture, or the difference between a benign polymorphism and a pathogenic variant at 0.01% allele frequency. Bioinformatics requires domain-specific reasoning that can weigh evidence according to established frameworks, not just pattern-match from training text.
Transparent methodology
Every result should come with a clear chain of evidence. Which databases were consulted? What criteria were applied? What code was executed? This is not just good practice. In clinical genomics, it is a regulatory expectation. Researchers need to verify AI-generated conclusions, not accept them on faith.
Execution capability
As discussed above, generating code is not the same as running it. A platform that suggests analyses but cannot execute them just shifts the bottleneck from “writing the script” to “running the script.” End-to-end execution in containerized environments, with appropriate compute resources, is what makes no-code bioinformatics actually work.
Flexibility for technical users
The best accessible platforms do not force a binary choice between “no code” and “full code.” A researcher who starts with natural language queries should be able to inspect the generated code, modify it, and run custom analyses when their needs go beyond what the interface can express. This prevents the platform from becoming a black box and ensures it scales with the researcher’s growing expertise.
The Broader Impact on Research
Making bioinformatics accessible to non-programmers is not just a convenience improvement. It has structural implications for how biological research gets done.
Faster hypothesis testing
When a researcher can go from question to answer in minutes instead of days, the feedback loop tightens. This means more hypotheses get tested, dead ends are identified sooner, and productive lines of inquiry are pursued faster. The rate of scientific iteration increases.
Democratized access
Currently, the quality of computational analysis a researcher can perform correlates strongly with their institution’s bioinformatics resources. Well-funded academic medical centers and large pharma companies have dedicated bioinformatics teams. Smaller labs, community hospitals, and researchers in low-resource settings often do not. AI-powered platforms that require no coding expertise can help level this playing field.
Reduced bottlenecks
In many research groups, a single bioinformatician supports five or ten wet lab scientists. This creates a permanent backlog. When routine analyses can be handled through natural language interfaces, the bioinformatician’s time is freed for genuinely novel computational work, while the biologists get their results without waiting.
Better reproducibility
Standardized, documented, citation-backed analyses are inherently more reproducible than ad-hoc scripts written differently by each analyst. When the platform logs every query, database version, and parameter setting, the analysis is reproducible by default.
What This Does Not Replace
It is worth being explicit about what no-code bioinformatics does not do.
It does not replace bioinformaticians. Novel method development, custom pipeline engineering, large-scale data processing infrastructure, and computational biology research all require deep programming expertise. AI-powered platforms handle routine and semi-routine analyses. They do not invent new algorithms.
It does not replace expert judgment. An AI can present evidence and apply frameworks, but the final interpretation, especially in clinical settings, requires human expertise. A pathogenic variant classification suggested by an AI still needs review by a trained geneticist. A drug target identified through computational analysis still needs experimental validation.
It does not eliminate the need to understand biology. A natural language interface makes computation accessible, but the researcher still needs to know what questions to ask, how to evaluate the answers, and what the biological implications are. Domain expertise remains essential.
Where the Field Is Heading
The trajectory is clear. As AI for biology matures, the interface between researchers and computational tools will continue to shift from programming languages to natural language. This does not mean code disappears. It means code becomes infrastructure rather than a prerequisite.
Several trends are accelerating this shift:
Multimodal analysis. Platforms are beginning to handle not just text-based queries but also direct data uploads: VCF files, FASTQ files, expression matrices. The natural language layer sits on top of automated data processing pipelines.
Agentic workflows. Instead of answering single questions, AI systems are beginning to plan and execute multi-step analyses autonomously. A researcher describes an end goal, and the system determines the sequence of analyses needed to get there.
Collaborative features. Shared workspaces where biologists and bioinformaticians can work together, with the AI handling translation between natural language descriptions and computational implementations, are beginning to appear.
Integration depth. The number of databases and tools connected to these platforms continues to grow. The more comprehensive the integration, the more complete the analysis a non-programmer can obtain through a single query.
Making the Shift
For researchers considering this transition, the practical advice is straightforward. Start with analyses you currently do manually across multiple databases. Variant interpretation, gene-disease lookups, and protein function queries are natural starting points. These are well-defined tasks where natural language interfaces can demonstrate immediate value.
If you work in a clinical setting, look for platforms that provide auditable evidence chains. In research, prioritize platforms that allow you to inspect and export the underlying code and data. In either case, verify results against databases you trust until you are confident in the platform’s accuracy.
The goal is not to stop learning computational skills. Researchers who understand the basics of what is happening under the hood, even if they never write a script, will ask better questions and evaluate answers more critically. The point is that computational literacy should not be a prerequisite for computational analysis.
Bioinformatics without coding is not about making biology simpler. It is about making computational biology as accessible as the questions biologists already know how to ask.
To explore how a molecular intelligence platform approaches this problem, with natural language queries, containerized code execution, and 30+ integrated databases, visit purna.ai. Researchers can apply for up to $10,000 in free research credits to test these workflows on their own data.
MIP is Purna AI’s Molecular Intelligence Platform, an AI-powered workspace for biology teams. Natural language bioinformatics, variant interpretation, protein structure prediction, code execution in containerized environments, and 30+ database integrations in one place. Explore the platform at purna.ai.
Explore Purna's Molecular Intelligence Platform
AI-powered workspace for biology teams to accelerate drug discovery from target identification to lead optimization.
Try Purna AI →
