How to Query Biological Databases Using Natural Language

Every researcher who works with genomic or proteomic data knows the routine. You start with a variant or gene of interest, then open ClinVar in one tab, gnomAD in another, UniProt in a third, OMIM in a fourth, and maybe PDB and PharmGKB after that. Each database has its own search syntax, its own identifiers, and its own way of presenting results. By the time you have stitched together a complete picture, 30 minutes have passed and you have barely started on your actual analysis. Natural language biological database query changes this workflow fundamentally, replacing six browser tabs and manual cross-referencing with a single conversational question.
This post walks through what it looks like in practice: real queries for variant lookup, gene-disease associations, protein structure retrieval, and drug-gene interactions. For each example, we contrast the traditional multi-tool workflow with a single natural language query to show where the time savings and integration benefits are most significant.
Why Biological Database Queries Are Still Painful
The problem is not that biological databases are poorly built. ClinVar, gnomAD, UniProt, OMIM, PDB, PharmGKB, and LOVD are each excellent at what they do. The problem is that biological questions rarely live within the boundaries of a single database.
A clinical geneticist evaluating a variant of uncertain significance (VUS) needs to check its classification in ClinVar, its population frequency in gnomAD, the associated gene’s disease links in OMIM, the protein’s functional domains in UniProt, and potentially the 3D structural context from PDB. A pharmacogenomics researcher investigating a drug-gene interaction needs PharmGKB for the clinical annotation, ClinVar for variant pathogenicity, and gnomAD for allele frequencies across populations.
Each of these databases uses different identifiers (rs numbers, HGVS notation, gene symbols, UniProt accession IDs, PDB codes), different search interfaces, and different output formats. Translating between them is manual, error-prone, and slow.
A 2023 survey published in Briefings in Bioinformatics found that clinical genomics researchers spend an average of 30-45 minutes per variant on manual database cross-referencing. For labs processing hundreds of variants per week, this adds up to a significant bottleneck that delays reporting and increases the risk of overlooking relevant evidence.
How Natural Language Biological Database Query Works
The concept is straightforward: instead of learning the query syntax for each individual database, you ask a question in plain English. The system parses your intent, identifies which databases are relevant, queries them in parallel, synthesizes the results, and returns a unified answer with citations pointing back to each source.
This is not the same as asking a general-purpose large language model (LLM) about biology. General LLMs draw on their training data, which may be months or years out of date, and they cannot distinguish between what they “know” and what they are fabricating. A natural language database query system pulls live data from the actual databases at query time, which means the results reflect the current state of each resource and every claim is traceable to a specific database entry.
The distinction matters. When ClinVar reclassifies a variant from VUS to Pathogenic, a live query system reflects that change immediately. A general LLM trained six months ago does not.
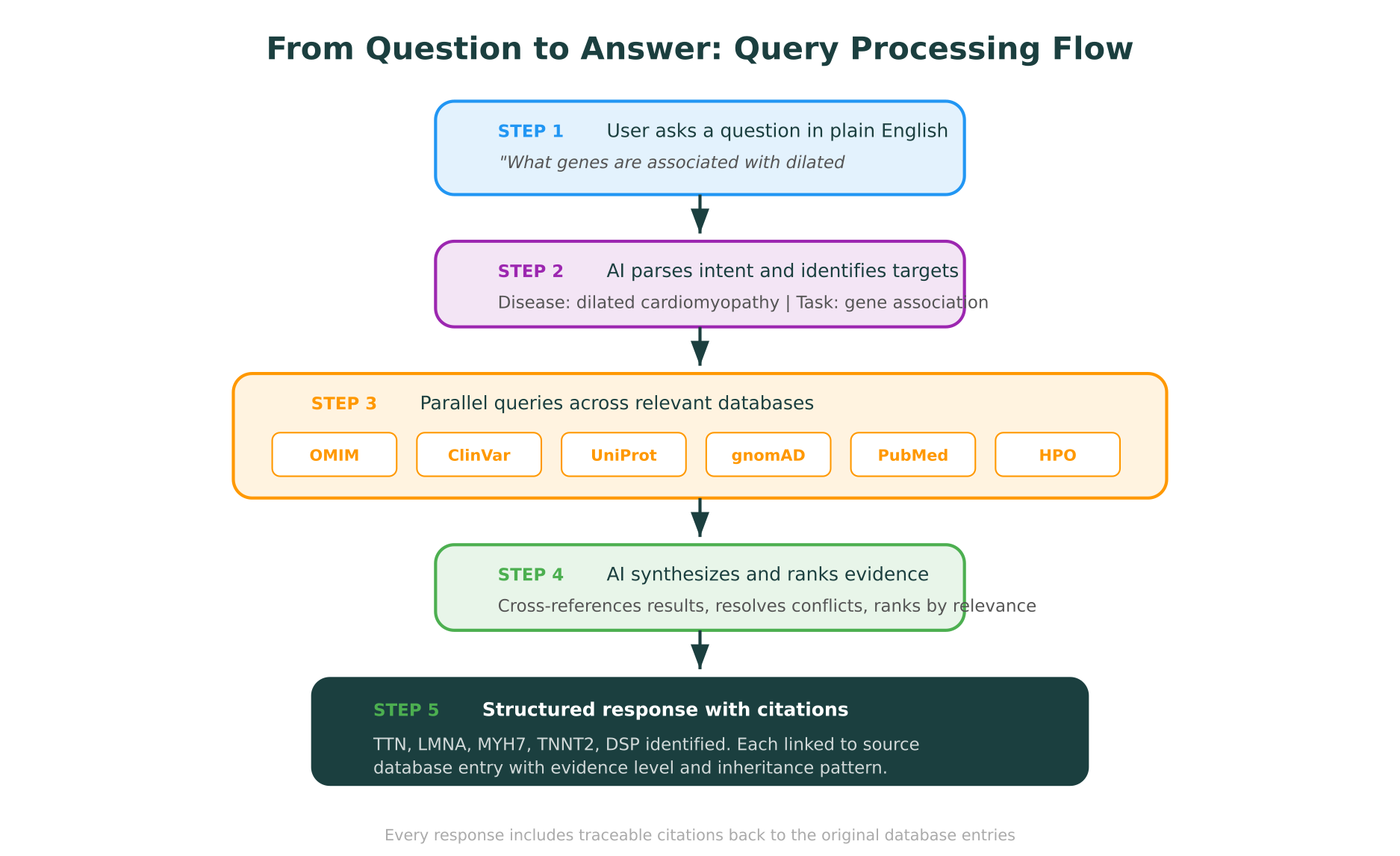
The query processing pipeline
When you type a natural language question into a system like Purna’s Molecular Intelligence Platform (MIP), the processing follows a structured pipeline:
-
Intent parsing: The AI identifies what you are asking for (variant classification, gene-disease association, protein structure, drug interaction) and extracts the relevant biological entities (gene names, variant notation, disease terms).
-
Database routing: Based on the intent, the system determines which of its 30+ integrated databases to query. A variant classification question routes to ClinVar, gnomAD, and LOVD. A protein structure question routes to PDB, AlphaFold, and UniProt.
-
Parallel querying: The system queries all relevant databases simultaneously, translating your natural language into the appropriate API calls and search parameters for each one.
-
Evidence synthesis: Results from multiple databases are cross-referenced, conflicts are flagged, and the evidence is ranked by relevance and quality.
-
Cited response: You receive a structured answer where every factual claim links back to its source database entry, so you can verify anything with a single click.
Practical Example 1: Variant Lookup and Classification
The traditional workflow
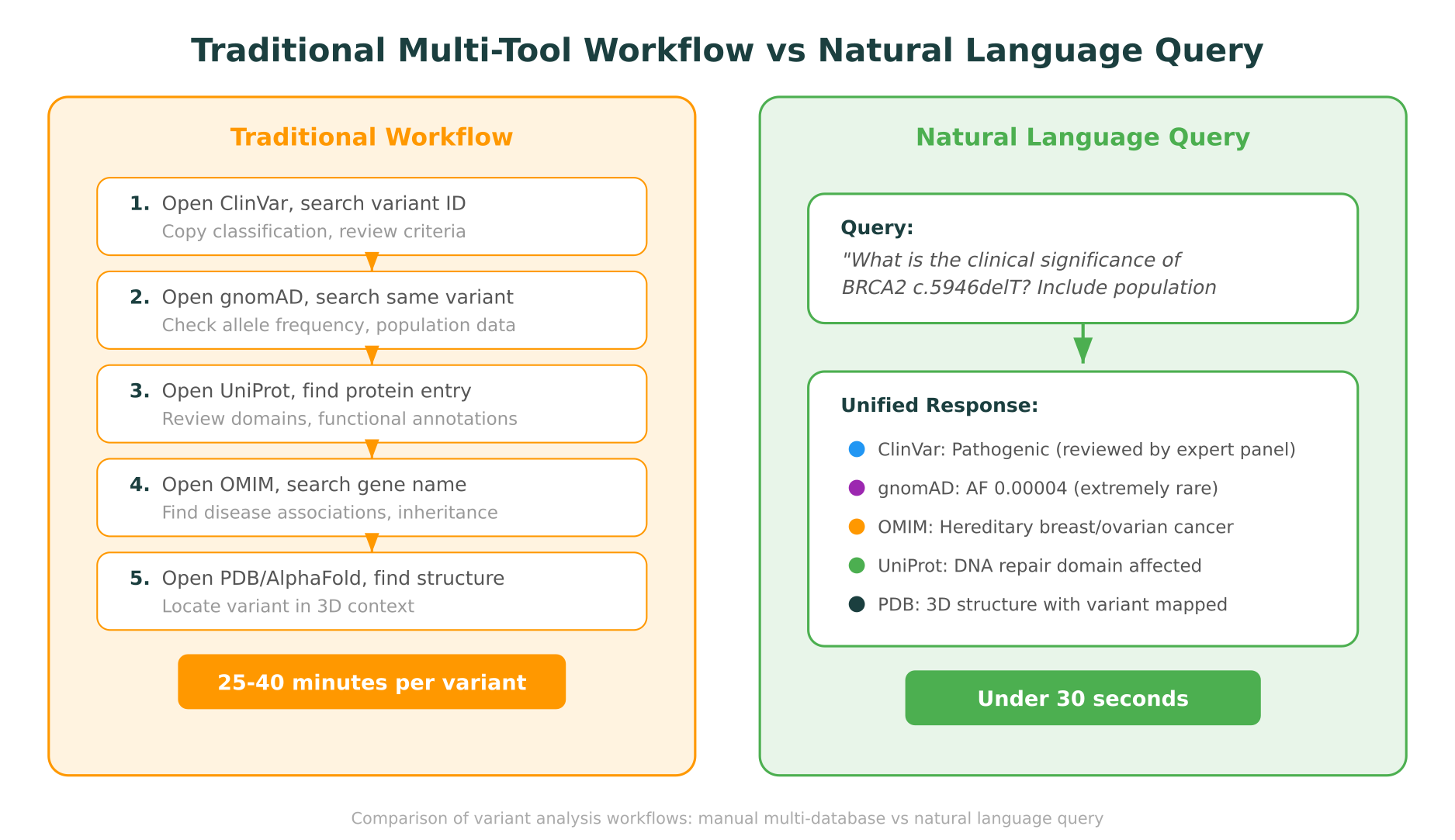
Suppose you encounter the variant BRCA2 c.5946delT in a patient’s exome sequencing results and need to determine its clinical significance.
Step 1: ClinVar (5-8 minutes). Navigate to ClinVar, search for “BRCA2 c.5946delT” or the corresponding rs number. Review the clinical significance classification, the number of submitters, the review status, and any condition associations. Note the variation ID for your records.
Step 2: gnomAD (3-5 minutes). Open gnomAD in a new tab. Search for the same variant using HGVS notation or genomic coordinates (you may need to convert between coordinate systems). Check the overall allele frequency, the population-specific frequencies, and whether the variant is absent from controls.
Step 3: OMIM (3-5 minutes). Search OMIM for BRCA2 to review the gene’s known disease associations, inheritance patterns, and any genotype-phenotype correlation data.
Step 4: UniProt (3-5 minutes). Find the BRCA2 protein entry in UniProt. Locate where the variant falls relative to known functional domains, binding regions, and post-translational modification sites.
Step 5: PDB/AlphaFold (5-10 minutes). Check whether an experimental structure exists in PDB that covers the region of interest. If not, retrieve the AlphaFold predicted structure. Identify where the variant maps in 3D space and whether it falls in a structurally critical region.
Total time: 25-40 minutes, assuming no identifier translation errors and no database downtime.
The natural language query
In MIP, the same task requires a single query:
“What is the clinical significance of BRCA2 c.5946delT? Include population frequency, disease associations, protein domain context, and structural impact.”
The response arrives in seconds and includes:
- ClinVar classification with review status, submitter count, and associated conditions, linked to the specific ClinVar entry
- gnomAD allele frequency across populations, with a note on whether the variant is absent from population controls
- OMIM disease associations for BRCA2, including hereditary breast-ovarian cancer syndrome with autosomal dominant inheritance
- UniProt domain mapping showing which functional region the variant falls in
- Structural context with the option to visualize the variant position in Molstar, MIP’s integrated 3D protein viewer
Every data point includes a citation to its source database. If ClinVar shows “Pathogenic (reviewed by expert panel)” while a single submitter has submitted it as “Likely pathogenic,” both pieces of information are presented transparently.
For teams processing large numbers of variants, this difference compounds. What used to take a full day of manual lookups for 20 variants can be completed in under an hour. This is particularly relevant for clinical labs working on variant interpretation at scale, where turnaround time directly affects patient care.
Practical Example 2: Gene-Disease Association Queries
The traditional workflow
A researcher studying dilated cardiomyopathy (DCM) wants to identify which genes are most strongly associated with the condition and what evidence supports each association.
Step 1: OMIM (5-8 minutes). Search for “dilated cardiomyopathy” in OMIM. Sift through multiple entries (DCM has over 20 listed genetic subtypes). Note the associated gene symbols and MIM numbers.
Step 2: ClinVar (5-10 minutes). For each gene identified in OMIM, search ClinVar to see how many pathogenic/likely pathogenic variants have been reported. This involves multiple individual searches.
Step 3: UniProt (5-8 minutes). For the top candidate genes, check UniProt for functional annotations, tissue expression data, and whether the protein is involved in cardiac muscle structure or function.
Step 4: PubMed (10-15 minutes). Search PubMed for recent reviews or meta-analyses linking each gene to DCM to assess the strength of the clinical evidence.
Total time: 25-40 minutes, and the result is a hand-compiled spreadsheet rather than a structured summary.
The natural language query
“Which genes are most strongly associated with dilated cardiomyopathy? Rank by evidence strength and include the number of pathogenic variants in ClinVar for each.”
MIP returns a structured response listing the top associated genes (TTN, LMNA, MYH7, TNNT2, DSP, and others), each with:
- OMIM disease association and inheritance pattern
- Number of pathogenic/likely pathogenic variants in ClinVar
- UniProt functional summary (sarcomere component, nuclear envelope protein, etc.)
- Key references from the literature
The response also flags that TTN truncating variants account for approximately 25% of familial DCM cases, which is a synthesis across databases that would take considerable manual effort to compile.
Practical Example 3: Protein Structure Retrieval and Analysis
The traditional workflow
A structural biologist wants to understand how the mutation TP53 R248W affects protein structure and function.
Step 1: UniProt (3-5 minutes). Search for TP53, identify the protein’s domain architecture, and confirm that position 248 falls in the DNA-binding domain.
Step 2: PDB (5-10 minutes). Search for TP53 structures in PDB. Multiple entries exist (over 400 as of 2026). Filter to find structures that include the DNA-binding domain at sufficient resolution. Download or view the structure.
Step 3: Literature (5-10 minutes). Search PubMed for studies on R248W to understand the functional impact. This is one of the most studied TP53 mutations, so there is abundant literature, but sorting through it takes time.
Step 4: Stability prediction (10-15 minutes). If you want to assess the structural impact computationally, navigate to DynaMut2 or a similar tool, upload the structure, specify the mutation, and wait for the calculation to complete.
Total time: 25-40 minutes, and the structural visualization is disconnected from the functional and clinical data.
The natural language query
“Show me the structural impact of TP53 R248W. Include stability prediction, domain context, and clinical significance.”
MIP retrieves the relevant PDB structure, maps the R248W position in the integrated Molstar viewer, runs a stability analysis via DynaMut2 (calculating delta-delta-G), and presents the results alongside ClinVar classification and functional annotations from UniProt. The 3D visualization is interactive, so you can rotate, zoom, and highlight the mutation site in the context of the full protein structure.
For researchers working on protein structure and mutation analysis, this integration eliminates the friction of moving between disconnected tools and manually correlating structural data with clinical significance.
Practical Example 4: Drug-Gene Interaction Queries
The traditional workflow
A pharmacogenomics researcher wants to understand how CYP2D6 variants affect the metabolism of tamoxifen.
Step 1: PharmGKB (5-8 minutes). Search PharmGKB for the tamoxifen-CYP2D6 pair. Review the clinical annotation, evidence level, and dosing guidelines.
Step 2: ClinVar (5-8 minutes). Search for CYP2D6 variants classified as affecting drug response. Cross-reference with the specific alleles mentioned in PharmGKB.
Step 3: gnomAD (5-8 minutes). Check population frequencies for the relevant CYP2D6 alleles to understand how common they are across different ancestry groups.
Step 4: Literature (10-15 minutes). Search for recent clinical studies on tamoxifen dosing based on CYP2D6 metabolizer status.
Total time: 25-40 minutes.
The natural language query
“What is the pharmacogenomic relationship between CYP2D6 and tamoxifen? Include relevant variant frequencies and clinical dosing implications.”
MIP pulls the PharmGKB clinical annotation (Level 1A evidence for CYP2D6-tamoxifen), lists the key CYP2D6 alleles that affect metabolizer status, retrieves their population frequencies from gnomAD, and summarizes the CPIC dosing guideline recommendations. Each section is cited to its source.
This type of cross-database synthesis is where natural language querying provides the most value. The relationships between pharmacogenomic variants, drug metabolism, and clinical dosing span multiple databases that were not designed to talk to each other.
What Makes This Different from Asking ChatGPT
It is worth addressing this question directly, because many researchers have experimented with asking general-purpose LLMs about variants and genes. The differences are significant:
Live data vs. training data. MIP queries the actual databases at the time you ask. General LLMs respond based on their training data, which may be months old. ClinVar alone processes thousands of new submissions per month. A variant classified as VUS six months ago may now be Pathogenic.
Citations vs. assertions. Every claim in MIP’s response links to a specific database entry that you can verify. General LLMs present information without traceable sources, making it impossible to distinguish accurate information from hallucinated content.
Structured querying vs. pattern matching. MIP translates your question into actual API calls to each database. General LLMs generate text that resembles a database query response but is actually reconstructed from patterns in their training data.
Domain-specific reasoning. MIP applies structured frameworks like ACMG/AMP criteria for variant classification. It does not simply describe what pathogenic means; it evaluates the specific evidence for the variant in question against the established criteria.
This distinction is explored in more depth in our post on how AI is making computational biology accessible, where we discuss the difference between tools that automate analysis and tools that merely generate text about analysis.
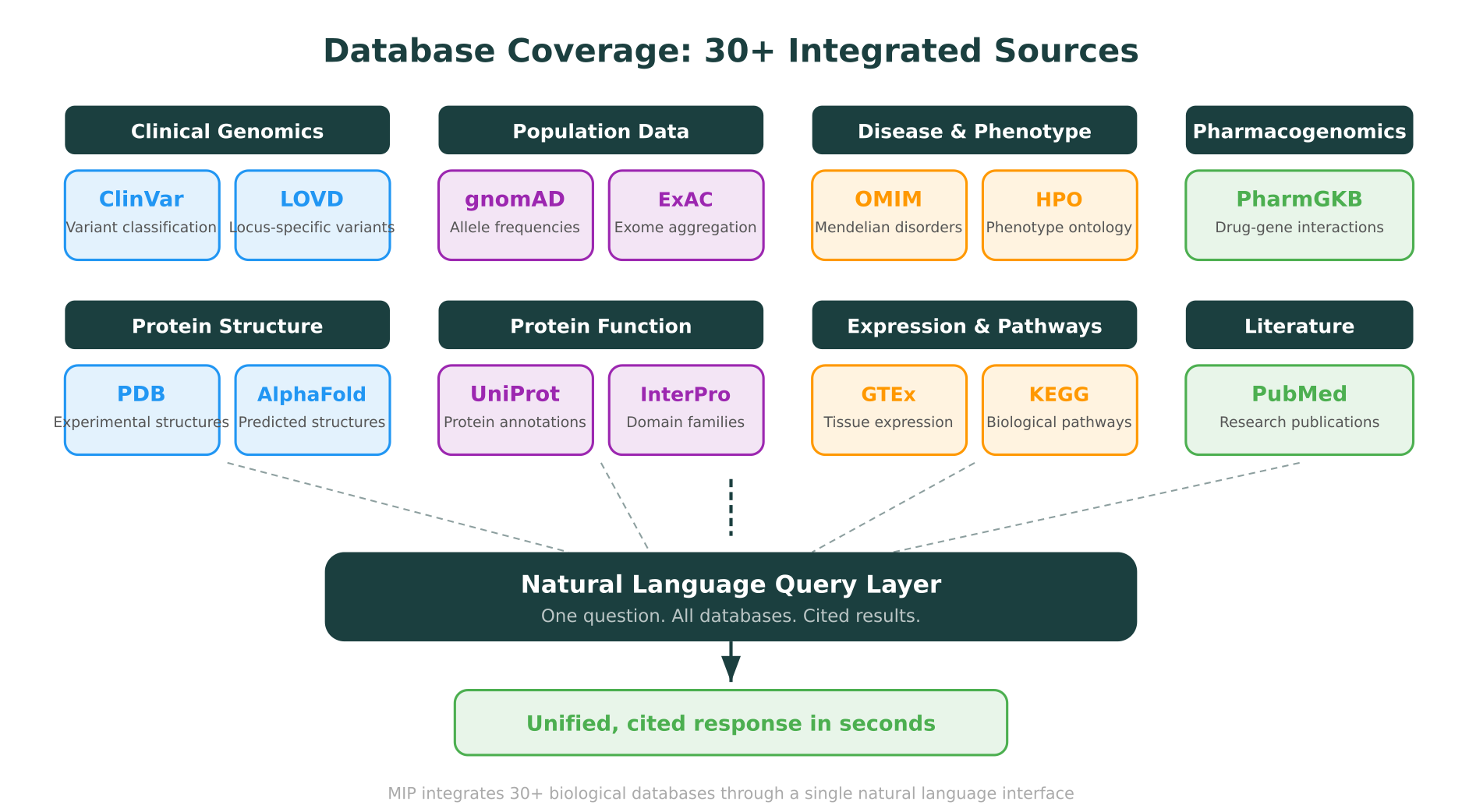
Databases You Can Query Through Natural Language in MIP
MIP integrates over 30 biological databases. Here are the ones most commonly queried through natural language, organized by category:
Clinical variant databases
- ClinVar: Variant-disease relationships, clinical significance classifications, submitter data, review status. Updated weekly.
- LOVD: Locus-specific variant databases maintained by expert curators. Particularly valuable for genes where ClinVar coverage is thin.
Population frequency databases
- gnomAD (v4): Allele frequencies across 807,162 individuals from diverse ancestries. Essential for filtering common variants and assessing population-specific carrier rates.
Disease and phenotype databases
- OMIM: Mendelian disease-gene relationships, inheritance patterns, clinical synopses.
- HPO (Human Phenotype Ontology): Standardized phenotype terms linked to genes and diseases. Useful for phenotype-driven gene prioritization.
Protein databases
- UniProt: Protein function annotations, domain architecture, post-translational modifications, subcellular localization.
- PDB: Experimentally determined 3D protein structures, rendered in MIP through the integrated Molstar viewer.
- AlphaFold Database: Predicted structures for proteins without experimental data.
- InterPro: Protein family and domain classifications.
Pharmacogenomics databases
- PharmGKB: Drug-gene relationships, clinical annotations, dosing guidelines, and pharmacogenomic variant data.
Functional and pathway databases
- GTEx: Tissue-specific gene expression data across 54 human tissues.
- KEGG: Biological pathway maps linking genes to metabolic and signaling networks.
Literature
- PubMed: Searchable from within MIP, with AI-assisted summarization of relevant publications.
The key point is that you do not need to know which database to query. When you ask “What is the population frequency of BRCA1 c.5266dupC across European and East Asian populations?”, MIP routes the query to gnomAD automatically. When you ask “Is there a known drug interaction for DPYD variants and fluorouracil?”, it queries PharmGKB without you specifying the database.
Unified Biological Database Search in Practice
The real benefit of natural language biological database query is not just speed. It is the ability to ask questions that span multiple databases in a single interaction.
Consider these examples of queries that would traditionally require visits to three or more databases:
Cross-database variant analysis: “Classify MEFV M694V according to ACMG criteria and show me population frequencies, protein domain impact, and whether any pharmacogenomic annotations exist.”
This single query triggers lookups in ClinVar (classification), gnomAD (population frequency), UniProt (domain mapping), PharmGKB (drug interactions), and applies ACMG/AMP framework reasoning across the combined evidence.
Comparative gene analysis: “Compare the pathogenic variant landscape of BRCA1 vs BRCA2. Which has more pathogenic variants in ClinVar? How do their protein domain architectures differ?”
Phenotype-to-gene: “A patient presents with hypertrophic cardiomyopathy, hearing loss, and short stature. What genes could explain this combination of phenotypes?”
This routes to OMIM and HPO for phenotype matching, then cross-references with ClinVar for variant data in the candidate genes.
Structure-informed interpretation: “I found a VUS at position 342 in KCNQ1. Show me the 3D structure, check if this position is in a conserved domain, and tell me whether nearby positions have known pathogenic variants.”
This combines PDB/AlphaFold (structure), UniProt/InterPro (domain conservation), and ClinVar (nearby pathogenic variants) into a single response with interactive 3D visualization.
These are the kinds of questions that researchers and clinicians actually ask. They do not think in terms of individual databases. They think in terms of biological questions. A natural language interface matches that cognitive model.
Limitations and Honest Caveats
Natural language database querying is not a replacement for expert judgment, and it is important to be transparent about its limitations.
Query ambiguity. Natural language is inherently ambiguous. “What is the significance of TP53?” could mean clinical significance of a specific variant, the biological function of the gene, or the gene’s role in cancer. Good systems handle this by asking clarifying questions or providing a comprehensive response that covers multiple interpretations. But edge cases exist.
Database coverage gaps. No single system integrates every biological database in existence. If your question requires data from a niche resource that is not integrated, you will still need to query it manually.
Novel variants. For truly novel variants with no database entries, a natural language query system can only report the absence of existing data. It can still provide supporting context (protein domain, conservation scores, nearby variants), but it cannot manufacture evidence that does not exist.
Computational analyses have limits. Stability predictions from tools like DynaMut2 are computational estimates, not experimental measurements. MIP presents them as such, with appropriate confidence indicators.
The goal is not to automate away the need for biological expertise. It is to eliminate the mechanical overhead of database navigation so that researchers can focus on interpretation and decision-making. As discussed in our overview of AI for biology in 2026, the most effective AI tools in the life sciences are those that augment expert workflows rather than attempting to replace them.
Getting Started with Natural Language Database Queries
If you spend a meaningful portion of your week querying ClinVar, gnomAD, UniProt, OMIM, or any of the other databases discussed in this post, natural language querying can reclaim hours of that time.
MIP is available to academic and clinical researchers through the research credits program, which provides up to $10,000 in compute credits for qualifying research projects. This makes it practical to explore natural language querying for your specific use case without upfront commitment.
The most effective way to evaluate the approach is to start with the queries you run most frequently. Take a variant you recently classified manually, ask MIP the same question, and compare the completeness, accuracy, and speed of the response against your manual workflow.
To explore the full capabilities of the Molecular Intelligence Platform, visit purna.ai.
Purna’s Molecular Intelligence Platform (MIP) integrates 30+ biological databases through a natural language interface, providing cited, live-data responses for variant interpretation, gene-disease analysis, protein structure queries, and pharmacogenomic lookups. Every answer traces back to its source database entry.
Explore Purna's Molecular Intelligence Platform
AI-powered workspace for biology teams to accelerate drug discovery from target identification to lead optimization.
Try Purna AI →
