Protein-Ligand Docking in the Age of AI: Classical vs Deep Learning Approaches

Protein-ligand docking has been a cornerstone of computational drug discovery for over three decades. The basic question it addresses is straightforward: given a protein target and a small molecule, where and how strongly does the molecule bind? But the tools available to answer that question have changed dramatically. Classical protein-ligand docking tools like AutoDock Vina, Glide, and GOLD rely on physics-based scoring functions and systematic conformational search. A new generation of AI-powered docking methods, including DiffDock, EquiBind, and Uni-Mol, approaches the same problem through learned representations and generative modeling. For drug discovery teams evaluating their computational toolkit in 2026, understanding the trade-offs between these approaches is essential.
This guide covers what each class of tools does well, where each falls short, how AI-predicted protein structures from tools like AlphaFold are reshaping the docking landscape, and practical recommendations for choosing the right approach for your project.
How Classical Molecular Docking Works
Classical molecular docking methods have been refined over decades and remain the most widely used approach in pharmaceutical R&D. These tools share a common architecture: a search algorithm that samples ligand conformations and orientations within a defined binding site, paired with a scoring function that estimates binding affinity.
Scoring functions
Classical docking tools use one of three types of scoring functions:
- Force-field-based scoring functions calculate interaction energies using molecular mechanics force fields (van der Waals, electrostatic, solvation). GOLD and HADDOCK fall into this category.
- Empirical scoring functions decompose binding free energy into a weighted sum of physically meaningful terms (hydrogen bonds, hydrophobic contacts, entropy loss) calibrated against experimental binding data. Glide SP and XP use this approach.
- Knowledge-based scoring functions derive statistical potentials from observed atom-pair frequencies in experimentally determined protein-ligand complexes. DrugScore and PMF are examples.
Each approach has known limitations. Force-field scoring tends to overestimate electrostatic contributions. Empirical functions depend heavily on the quality and diversity of their training set. Knowledge-based potentials can miss novel interaction geometries not well represented in the PDB.
Search algorithms
The search problem in docking is challenging: even a modest drug-like molecule with 5-10 rotatable bonds has millions of possible conformations in three-dimensional space. Classical tools handle this through genetic algorithms (GOLD), Monte Carlo simulated annealing (AutoDock 4), or exhaustive grid-based search with local optimization (AutoDock Vina, Glide).
AutoDock Vina, published in the Journal of Computational Chemistry in 2010, introduced a fast gradient-based optimization that made it possible to dock a single ligand in minutes on commodity hardware. It remains one of the most cited docking tools, with tens of thousands of users. Glide, developed by Schrodinger, offers a multi-stage funnel that progresses from rough shape matching (HTVS mode) to detailed pose refinement (XP mode), making it well suited for virtual screening campaigns of varying scales.
Strengths of classical approaches
Classical docking methods have several practical advantages that explain their continued dominance:
- Interpretability. Every score component maps to a physical interaction. Medicinal chemists can inspect a docking pose and understand why the scoring function ranked it highly.
- Reliability for known binding sites. When a co-crystal structure of the target with a related ligand exists, classical docking consistently reproduces the binding mode within 2 angstroms RMSD for well-behaved systems.
- Extensive validation. Decades of benchmarking studies provide clear guidance on when these tools work and when they do not.
- No GPU required. Most classical docking runs on CPUs, making it accessible to groups without specialized hardware.
- Regulatory acceptance. Pharmaceutical companies have extensive experience documenting classical docking results for regulatory submissions.
Limitations
The well-known limitations of classical docking include poor handling of protein flexibility (most tools treat the receptor as rigid or semi-rigid), inconsistent performance on novel binding sites, and scoring functions that correlate weakly with experimental binding affinities in many systems. Virtual screening enrichment, while consistently above random, often plateaus at modest levels, requiring large experimental follow-up campaigns.
AI-Based Protein-Ligand Docking Tools
Deep learning approaches to molecular docking reframe the problem entirely. Rather than sampling conformations and scoring them with physics-based functions, AI docking tools learn the mapping from protein-ligand inputs to binding poses directly from structural data.
DiffDock
DiffDock, published by Corso et al. in ICLR, 2023, applies diffusion generative modeling to molecular docking. The model learns to generate ligand poses by iteratively denoising from a random initial placement, conditioned on the protein structure. This approach has several notable properties:
- Blind docking capability. DiffDock does not require a predefined binding site. It predicts both the binding location and the ligand pose simultaneously, making it useful for targets without known binding pockets.
- Confidence scoring. The model outputs a confidence score for each generated pose, allowing users to assess prediction reliability.
- Speed. Inference takes seconds per ligand on a GPU, orders of magnitude faster than classical docking with comparable or better pose prediction accuracy on certain benchmarks.
DiffDock-L, a later iteration, expanded support to larger and more flexible ligands, addressing one of the initial model’s limitations. On the PoseBusters benchmark, which specifically tests for physically valid poses (no clashes, correct bond geometries), DiffDock’s performance improved substantially between its 2023 and 2025 versions.
However, DiffDock has important limitations. Its scoring confidence does not correlate well with binding affinity, meaning it predicts where a ligand binds more reliably than how strongly it binds. For virtual screening, where ranking compounds by predicted affinity is the goal, this is a significant gap.
EquiBind
EquiBind, from Stark et al., published in ICML, 2022, takes a different architectural approach. Rather than diffusion, it uses an SE(3)-equivariant graph neural network to directly predict the ligand’s bound pose in a single forward pass. This makes EquiBind extremely fast but less accurate than DiffDock for pose prediction. It is best suited as a rapid pre-filter in ultra-large-scale screening campaigns, where speed matters more than pose precision.
Uni-Mol Docking
Uni-Mol, developed by the DP Technology team, uses a 3D molecular pre-training framework that learns representations of both proteins and ligands. The docking module fine-tunes these representations for pose prediction. Uni-Mol’s strength is its generalizability: because the pre-trained representations capture broad chemical and structural knowledge, it handles diverse chemotypes more consistently than methods trained only on protein-ligand complexes.
NeuralPLexer
NeuralPLexer, published in Nature Machine Intelligence in 2024, represents an emerging hybrid approach. It jointly predicts protein conformational changes and ligand binding poses, effectively modeling induced-fit effects that classical docking handles poorly. For targets where binding involves significant protein reorganization, this approach can outperform both classical and other AI methods.
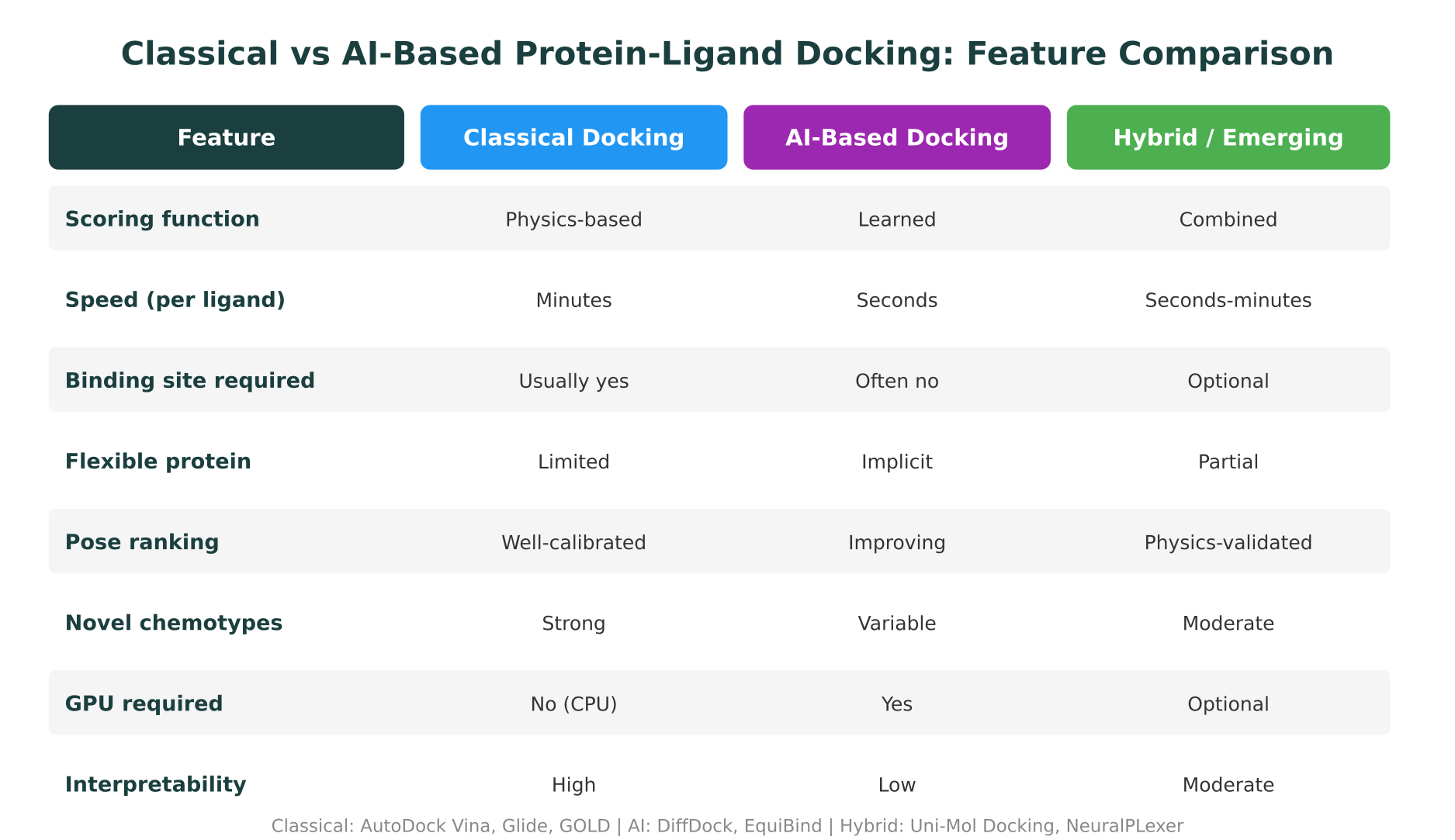
Head-to-Head: Classical vs AI Protein-Ligand Docking Tools
The table below summarizes the practical trade-offs across the most widely used tools in each category.
| Tool | Type | Speed | Binding Site Required | Protein Flexibility | Best Use Case |
|---|---|---|---|---|---|
| AutoDock Vina | Classical | Minutes/ligand | Yes | Limited (selected sidechains) | Academic screening, reproducible workflows |
| Glide (SP/XP) | Classical | Minutes/ligand | Yes | Limited | Pharma virtual screening, lead optimization |
| GOLD | Classical | Minutes/ligand | Yes | Selected sidechains | Pose prediction, metalloprotein targets |
| HADDOCK | Classical | Hours/complex | Optional (data-driven) | Moderate | Protein-protein and protein-ligand |
| DiffDock | AI | Seconds/ligand | No (blind docking) | Implicit | Blind docking, rapid exploration |
| EquiBind | AI | Milliseconds/ligand | No | None | Ultra-large library pre-filtering |
| Uni-Mol Docking | AI | Seconds/ligand | Optional | Implicit | Diverse chemotype screening |
| NeuralPLexer | AI/Hybrid | Seconds/complex | No | Joint prediction | Induced-fit targets |
Several patterns emerge from this comparison. Classical tools offer better interpretability and more reliable affinity ranking but require a known binding site and handle protein flexibility poorly. AI tools are faster and can perform blind docking but currently lack the scoring accuracy needed for late-stage lead optimization.
How AlphaFold Structures Are Changing the Docking Landscape
The availability of AI-predicted protein structures from AlphaFold, Boltz-2, and ESMFold has fundamentally expanded which targets are accessible to docking. Before 2021, docking was limited to targets with experimental crystal or cryo-EM structures, roughly 200,000 proteins in the PDB. The AlphaFold Protein Structure Database now provides predicted structures for over 200 million proteins, opening up previously undruggable targets to computational screening.
But using predicted structures for docking introduces specific challenges that drug discovery teams need to account for.
The binding site quality problem
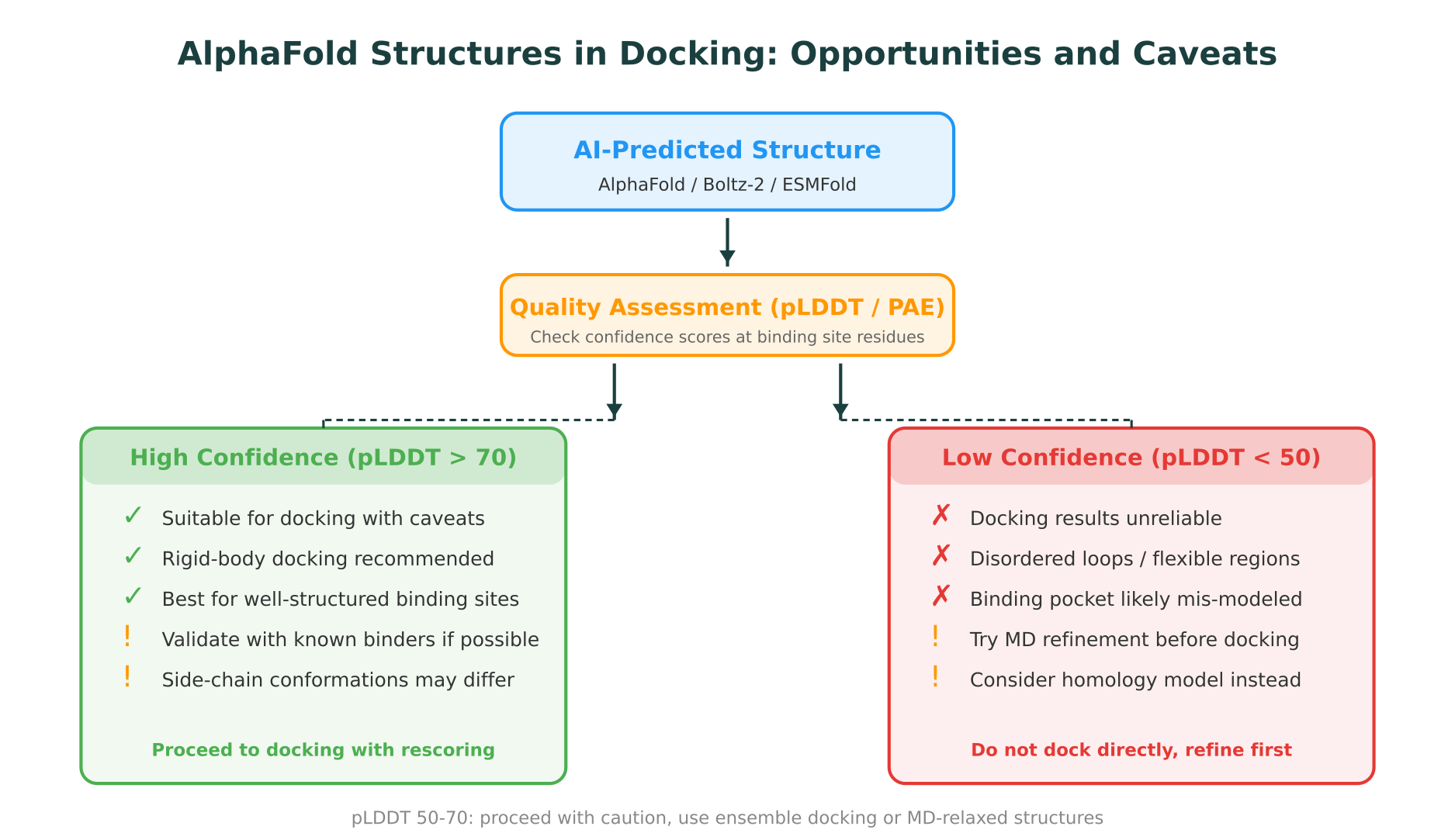
The accuracy of a docking result depends heavily on the quality of the binding site geometry. Experimental crystal structures, especially those with a co-crystallized ligand, capture the precise arrangement of side chains that form the binding pocket. AI-predicted structures get the overall fold correct for high-confidence regions (pLDDT > 70) but may not reproduce the exact side-chain rotamers that define a binding pocket. A study by Heo and Feig, published in Proteins, 2022, showed that docking into AlphaFold structures yields lower enrichment compared to crystal structures, primarily due to side-chain misplacement in the binding site.
Practical guidelines for docking into predicted structures
- Check per-residue confidence. If binding site residues have pLDDT scores below 50, docking results will likely be unreliable. For scores between 50-70, proceed with caution and use ensemble approaches.
- Relax the structure. Running a short molecular dynamics simulation (10-50 ns) or energy minimization before docking can improve side-chain positioning in the binding pocket.
- Use ensemble docking. Generate multiple conformations of the predicted structure (through MD or normal mode analysis) and dock into all of them. This partially compensates for the single-conformation limitation.
- Validate with known binders. If any experimental binding data exists for the target, dock known active and inactive compounds first to calibrate enrichment.
- Consider AI docking tools. DiffDock and NeuralPLexer were trained on both experimental and predicted structures, making them more tolerant of the structural noise present in AlphaFold models.
Teams working with predicted structures can benefit from platforms that integrate structure prediction with 3D visualization and stability analysis. Inspecting the binding pocket in a structure viewer, checking conservation of key residues, and running stability predictions for binding-site mutations all help assess whether a predicted structure is reliable enough for docking.
Practical Decision Guide: Which Approach Should You Use?
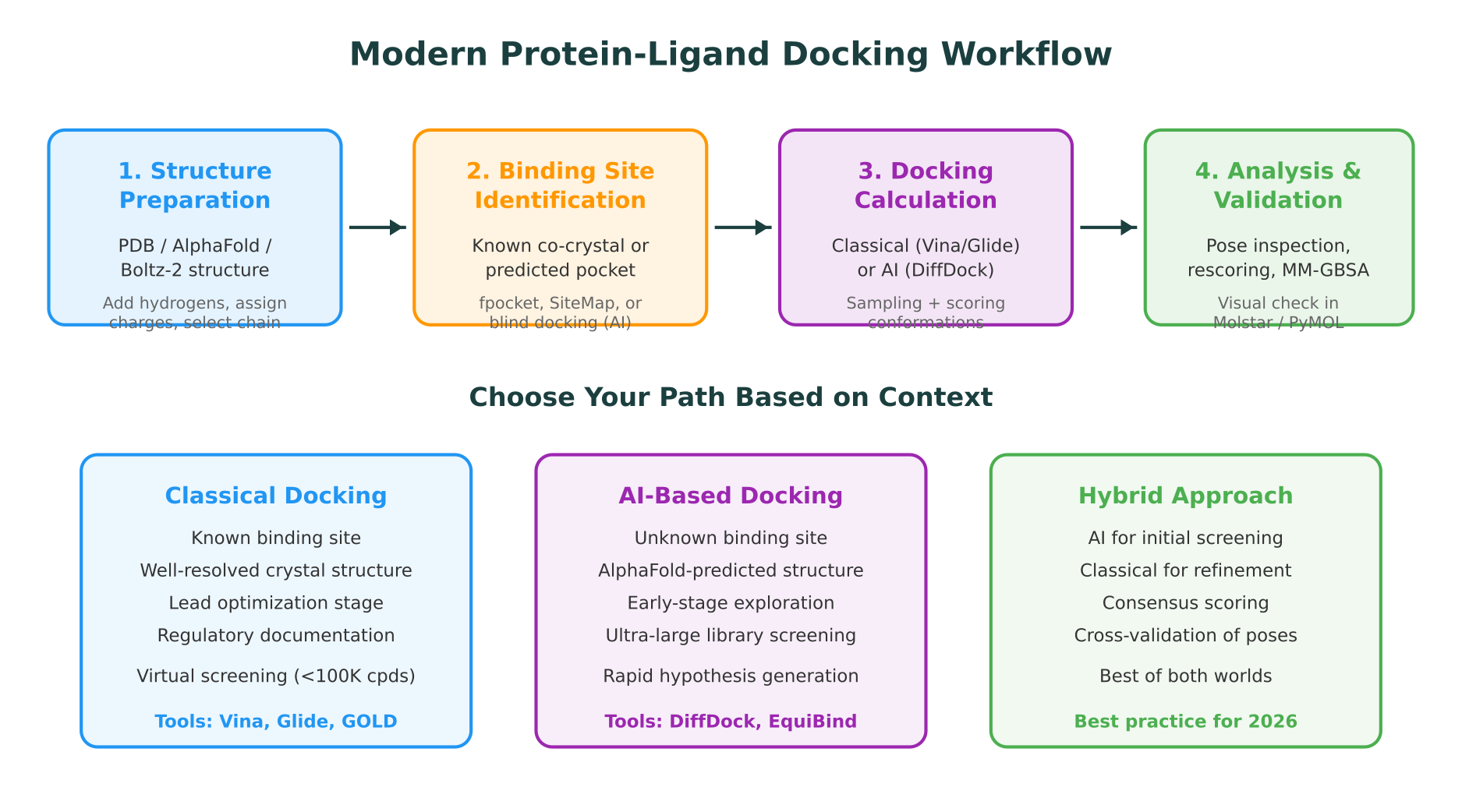
The right docking strategy depends on your project stage, data availability, and what question you are trying to answer.
Use classical docking when:
- You have a high-resolution crystal structure with a co-crystallized ligand
- You need reliable affinity ranking for lead optimization
- Your screening library is under 100,000 compounds
- Results need to be interpretable for medicinal chemistry decision-making
- You are preparing documentation for regulatory submissions
Use AI docking when:
- No experimental structure exists (only AlphaFold or Boltz-2 predictions)
- The binding site is unknown and you need blind docking
- You are screening ultra-large libraries (millions to billions of compounds)
- Speed is the primary constraint
- You are in early-stage target exploration, not lead optimization
Use a hybrid approach when:
- You want to combine the speed of AI for initial screening with the reliability of classical docking for refinement
- Multiple binding sites are possible and you need to narrow down candidates before detailed analysis
- You have predicted structures and want to cross-validate poses across methods
The hybrid approach is increasingly becoming the standard in industrial drug discovery pipelines. A typical workflow uses DiffDock or EquiBind to screen millions of compounds in hours, then re-docks the top 1,000-10,000 hits with Glide XP or GOLD for detailed pose analysis and affinity ranking.
Benchmarks and What They Tell Us
Several benchmarking studies have compared classical and AI docking methods head-to-head. The most informative include:
PoseBusters Benchmark (Buttenschoen et al., Chemical Science, 2024). This benchmark specifically tests whether predicted poses are physically valid: no steric clashes, correct bond lengths and angles, proper stereochemistry. DiffDock v1 initially showed high pose prediction rates but many physically invalid poses. Later versions and classical tools like Glide performed better on physical validity.
CASF-2016 (Su et al., Journal of Chemical Information and Modeling, 2019). The standard benchmark for scoring, ranking, docking, and screening power. Classical tools (Glide, GOLD) generally lead on scoring and ranking power. AI tools show competitive or superior docking power (pose prediction accuracy).
Merck FEP Benchmark (Schindler et al., Journal of Chemical Information and Modeling, 2020). Focused on relative binding free energy prediction for congeneric series. This benchmark highlights where classical free energy perturbation methods still significantly outperform AI docking for affinity prediction in lead optimization.
The overall picture: AI docking tools have largely caught up with classical methods for pose prediction (predicting where a ligand binds) but still lag behind for affinity prediction (predicting how strongly it binds). This distinction matters enormously depending on your application.
The Role of Protein Structure Visualization
Regardless of which docking method you use, visual inspection of predicted poses remains an essential quality control step. No scoring function, classical or learned, is reliable enough to skip manual review of top-ranked poses.
Molecular visualization tools allow researchers to check for chemically reasonable interactions (hydrogen bonds, pi-stacking, hydrophobic contacts), identify potential clashes, and assess whether the predicted pose makes sense given what is known about the target biology. As discussed in our comparison of PyMOL, ChimeraX, and Molstar, the choice of visualization tool matters for efficient analysis of docking results.
For teams that run docking as part of a broader structural biology workflow, having visualization integrated with protein structure prediction, stability analysis, and database lookups in a single environment reduces the friction of switching between disconnected tools. This is one area where a molecular intelligence platform can significantly accelerate the analysis cycle.
What Is Coming Next
Several trends are shaping the near-term future of protein-ligand docking:
Diffusion models for flexible docking. NeuralPLexer demonstrated that jointly predicting protein and ligand conformations is feasible. Expect more tools to handle induced-fit docking through generative modeling rather than expensive MD simulations.
Foundation models for molecular interactions. Large pre-trained models that understand both protein and small molecule chemistry (like Uni-Mol) are becoming general-purpose tools for predicting interactions, properties, and binding. These models are trained on far more data than any classical scoring function.
Integration with generative chemistry. AI docking is increasingly coupled with generative molecular design: a generative model proposes new molecules, an AI docking model predicts their binding poses, and the cycle repeats. This design-dock-iterate loop runs orders of magnitude faster than classical approaches.
Better confidence calibration. The gap between pose prediction and affinity prediction is the most important open problem. Improved confidence calibration, where the model’s predicted score correlates with experimental binding affinity, would make AI docking viable for lead optimization.
For teams in drug discovery who already work with AI-driven structural biology, adding AI docking to the workflow is a natural next step. The key is understanding which tool fits which question, and being realistic about what current methods can and cannot do.
Getting Started
For researchers looking to incorporate AI docking into their workflows, the barrier to entry has dropped significantly. DiffDock is available as open-source code on GitHub. AutoDock Vina remains freely available for academic use. Uni-Mol provides pre-trained models that can be fine-tuned on proprietary datasets.
The more challenging aspect is the surrounding infrastructure: preparing structures, managing ligand libraries, running calculations at scale, visualizing results, and connecting docking outputs to downstream analyses like stability prediction and database queries.
Purna’s Molecular Intelligence Platform provides an integrated environment where teams can pull protein structures from the PDB or AlphaFold, visualize binding sites in Molstar, run stability analysis with DynaMut2, and connect results to 30+ clinical and biological databases. For drug discovery teams who need to move from structure to hypothesis quickly, having these capabilities in one workspace removes the overhead of stitching together disconnected command-line tools.
Researchers can apply for up to $10,000 in free MIP credits to explore these workflows on Purna’s platform.
Purna AI’s Molecular Intelligence Platform (MIP) is an AI-powered workspace for biology teams. It brings together molecular analysis, variant interpretation, protein structure prediction, and clinical database integrations into one environment. Built for teams who work with biological data and need consistent, reproducible answers without juggling disconnected tools. Learn more at purna.ai.
Explore Purna's Molecular Intelligence Platform
AI-powered workspace for biology teams to accelerate drug discovery from target identification to lead optimization.
Try Purna AI →
