How AI Is Compressing Drug Discovery Timelines

Bringing a new drug to market takes, on average, 12 to 15 years and costs between $1 billion and $2 billion. Roughly 90% of drug candidates that enter clinical trials never reach approval. These numbers have barely changed in two decades, despite billions of dollars invested in automation, high-throughput screening, and computational chemistry.
AI is now compressing specific phases of this pipeline significantly, but the compression is unevenly distributed. The early stages of discovery, target identification, hit finding, and lead optimization, are where AI delivers the most dramatic time savings. Clinical trials, which account for the majority of both time and cost, remain largely unaffected by current AI capabilities.
This post breaks down where time is actually being saved, what the real-world evidence looks like as of early 2026, and where the honest limits remain.
The Traditional Drug Discovery Timeline
Before examining what AI changes, it is worth understanding the baseline. A traditional small-molecule drug program proceeds through a well-defined sequence of phases, each with its own timescale and failure modes.
Target identification: 1 to 2 years
The process begins with identifying a biological target, typically a protein, that plays a causal role in a disease. This involves mining published literature, analyzing omics datasets (genomics, transcriptomics, proteomics), and validating hypotheses through wet-lab experiments. A research team might spend 12 to 24 months reviewing evidence from databases like ClinVar, OMIM, and Open Targets before committing to a target.
Hit finding: 1 to 2 years
Once a target is validated, the next step is finding chemical compounds (“hits”) that modulate it. Traditional approaches include high-throughput screening (HTS) of compound libraries (often 1 to 2 million compounds) and fragment-based screening. Physical HTS campaigns are expensive and slow, requiring specialized equipment and months of optimization.
Lead optimization: 1 to 2 years
Initial hits rarely have the right combination of potency, selectivity, metabolic stability, and safety. Lead optimization involves iterative cycles of chemical modification, synthesis, and testing, the classic “design-make-test-analyze” (DMTA) loop. Each cycle takes weeks. Most programs require dozens of cycles to arrive at a clinical candidate.
Preclinical development: 1 to 2 years
Before a candidate can enter human trials, it must pass preclinical safety assessments. This includes in vitro toxicology, pharmacokinetic studies, formulation development, and animal studies (typically in two species). Regulatory agencies require specific data packages that cannot be shortcut.
Clinical trials: 6 to 10 years
Clinical development is the longest and most expensive phase. Phase I trials (safety in healthy volunteers) take 1 to 2 years. Phase II trials (efficacy in patients) take 2 to 3 years. Phase III trials (large-scale confirmation) take 3 to 5 years. Patient recruitment alone accounts for a significant fraction of this time, and regulatory review adds additional months.
Total: 12 to 15 years, $1 to $2 billion, with approximately a 90% failure rate in clinical trials.
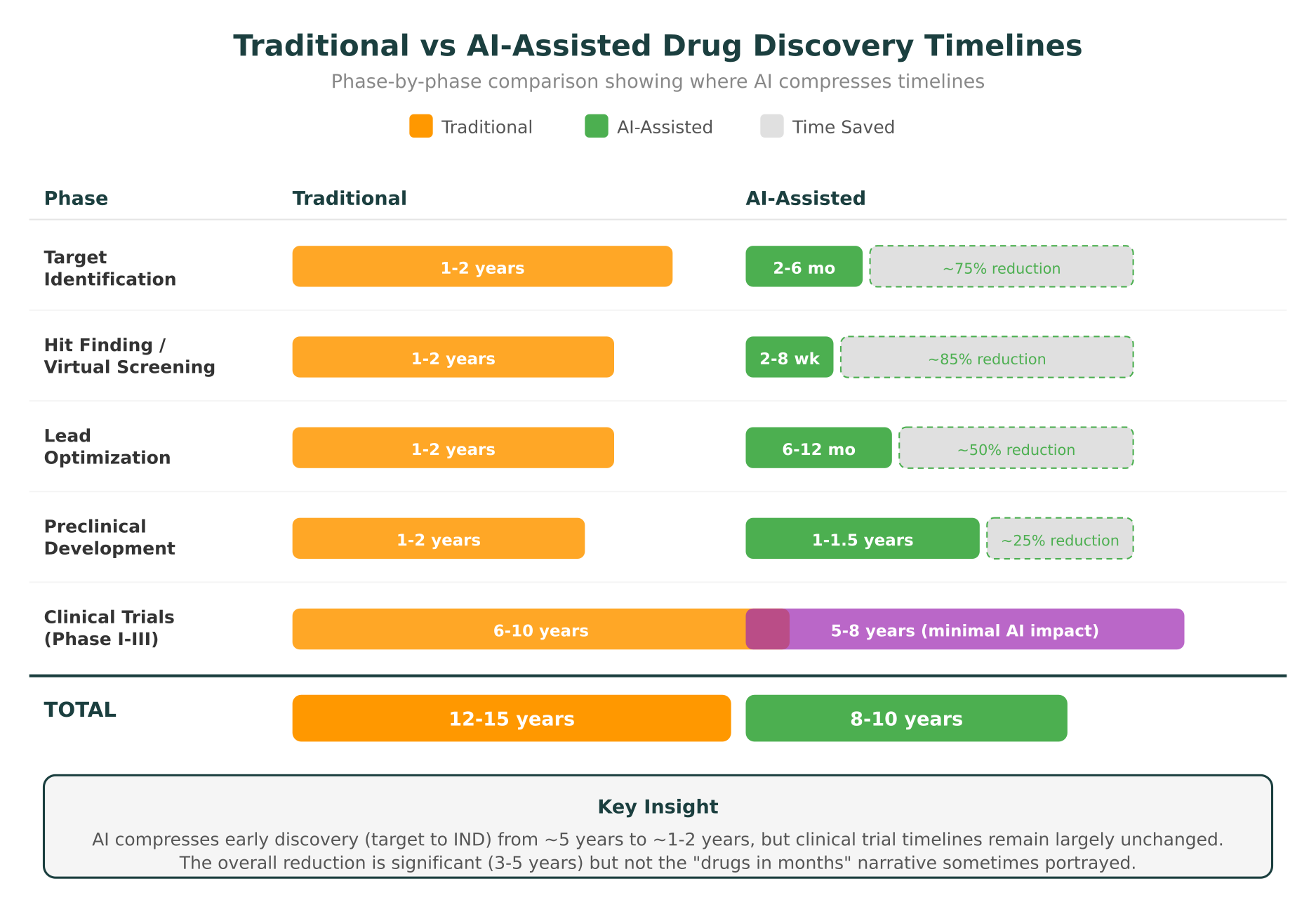
Where AI Is Actually Saving Time
The impact of AI on drug discovery timelines is concentrated in the first three phases: target identification, hit finding, and lead optimization. Together, these phases traditionally take 3 to 6 years. AI is compressing them to 1 to 2 years in well-executed programs.
Target identification: from years to months
AI’s impact on target identification comes from three capabilities: automated multi-omics integration, knowledge graph reasoning, and literature mining at scale.
Multi-omics integration. Platforms like Open Targets aggregate evidence from GWAS, expression data, protein interactions, and clinical annotations to produce composite target-disease association scores. What previously required a research team months of manual curation across dozens of databases can now be accomplished in days. Teams working with computational drug target discovery workflows can systematically rank hundreds of potential targets against multiple evidence types simultaneously.
Knowledge graphs. Companies like BenevolentAI and Recursion have built large-scale knowledge graphs that connect genes, proteins, pathways, diseases, and drugs. These graphs enable researchers to identify non-obvious connections between a disease and potential targets. For example, BenevolentAI’s knowledge graph identified baricitinib as a potential COVID-19 treatment by linking the drug’s known mechanism to the viral entry pathway, a connection that would have been extremely difficult to find through manual literature review.
Literature mining. Natural language processing models can now extract structured relationships from millions of biomedical publications. Rather than reading papers one at a time, researchers can query the entire corpus for specific relationships (e.g., “proteins that interact with KRAS and are upregulated in pancreatic cancer”). This type of natural language querying across biological databases is one of the defining capabilities of molecular intelligence platforms.
Estimated time savings: 75% reduction. From 1 to 2 years down to 2 to 6 months.
Hit finding: from years to weeks
This is where AI delivers the most dramatic acceleration. Traditional HTS requires physical screening of large compound libraries. AI-powered virtual screening can evaluate billions of compounds computationally.
AI-powered docking. Tools like DiffDock, a diffusion-based generative model for molecular docking published at ICLR 2023, can predict protein-ligand binding poses in seconds rather than the minutes required by classical docking tools like AutoDock Vina. For large virtual screening campaigns, this speed advantage is transformative. A team running protein-ligand docking workflows can now screen ultra-large virtual libraries (billions of compounds) in days rather than months.
Generative molecular design. Rather than screening existing compounds, generative chemistry models can propose entirely new molecules optimized for desired properties. Diffusion models and variational autoencoders trained on chemical space can generate novel scaffolds that satisfy multiple constraints simultaneously: binding affinity, ADMET properties, and synthetic accessibility. This approach expands the search space beyond known chemical matter, potentially accessing regions of chemical space that no physical library covers.
ADMET prediction. Absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties determine whether a hit compound can become a drug. ML models trained on large ADMET datasets can predict these properties for virtual compounds before any synthesis occurs. This allows teams to filter out problematic compounds early, focusing synthesis efforts on the most promising candidates.
Estimated time savings: 85% reduction. From 1 to 2 years down to 2 to 8 weeks for initial hit identification.
Lead optimization: fewer cycles, faster convergence
Lead optimization has traditionally been the most iteration-heavy phase. Each DMTA cycle requires designing a new compound, synthesizing it (days to weeks), testing it (days), and analyzing the results. Most programs go through 20 to 50 cycles.
ML-guided structure-activity relationships (SAR). Machine learning models trained on a program’s own data can predict which chemical modifications will improve potency, selectivity, or metabolic stability. Instead of synthesizing 20 analogs and hoping a few improve on the lead, teams can computationally evaluate hundreds of modifications and synthesize only the most promising 3 to 5. This reduces the number of DMTA cycles by 50% or more in well-designed programs.
Property prediction. Multi-task neural networks can predict solubility, microsomal stability, hERG liability, and other key properties from molecular structure alone. These predictions are not perfect, but they are accurate enough to deprioritize compounds with obvious liabilities before committing synthesis resources.
Retrosynthesis planning. AI-powered retrosynthesis tools (such as those from PostEra and IBM RXN) can propose synthetic routes for novel compounds in seconds. This accelerates the “make” step of the DMTA cycle by reducing the time chemists spend planning synthesis.
Understanding how protein mutations impact drug binding is also critical during lead optimization, particularly for biologic therapeutics and precision oncology programs where target variants may affect drug efficacy.
Estimated time savings: 50% reduction. From 1 to 2 years down to 6 to 12 months.
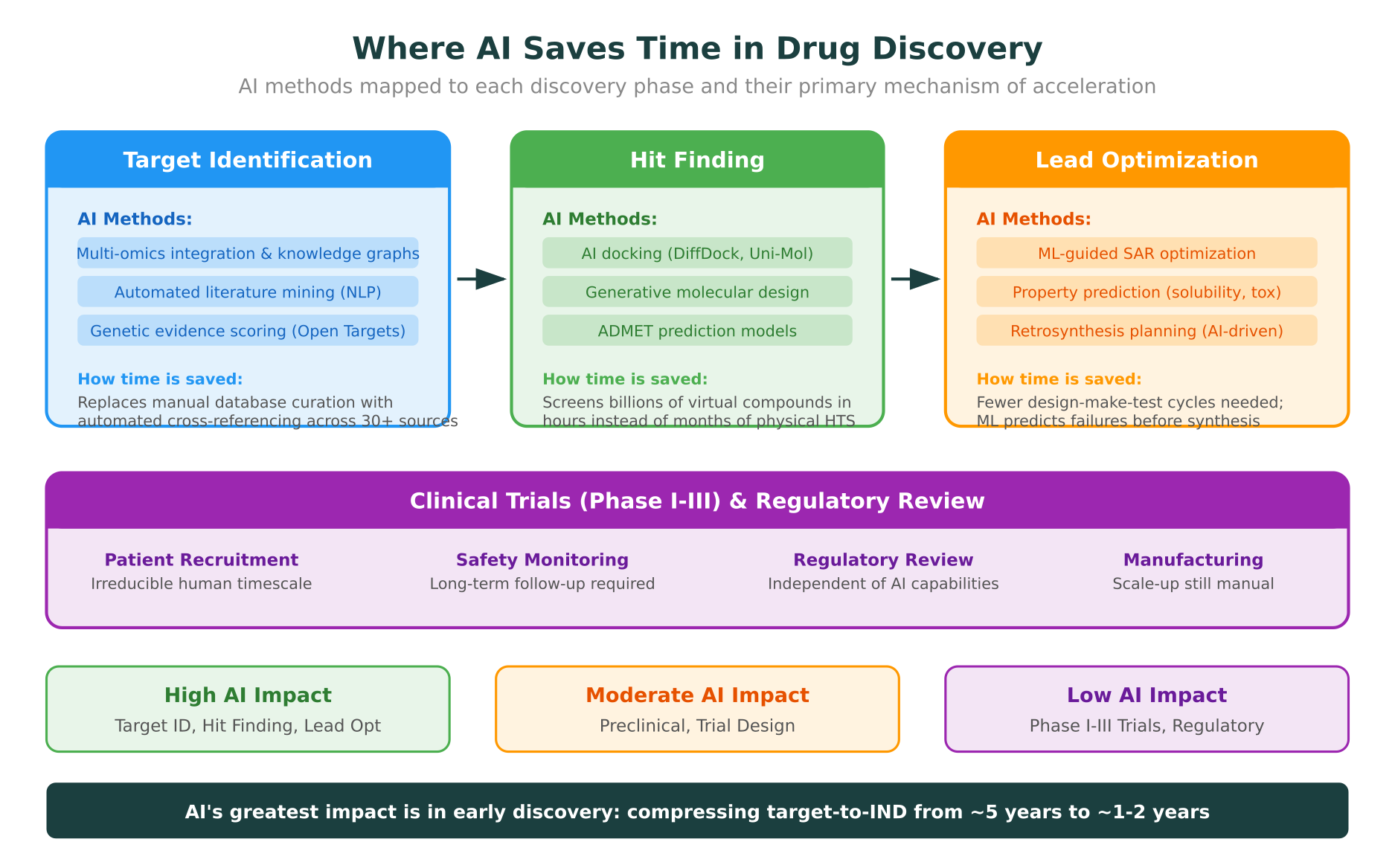
Real-World Examples
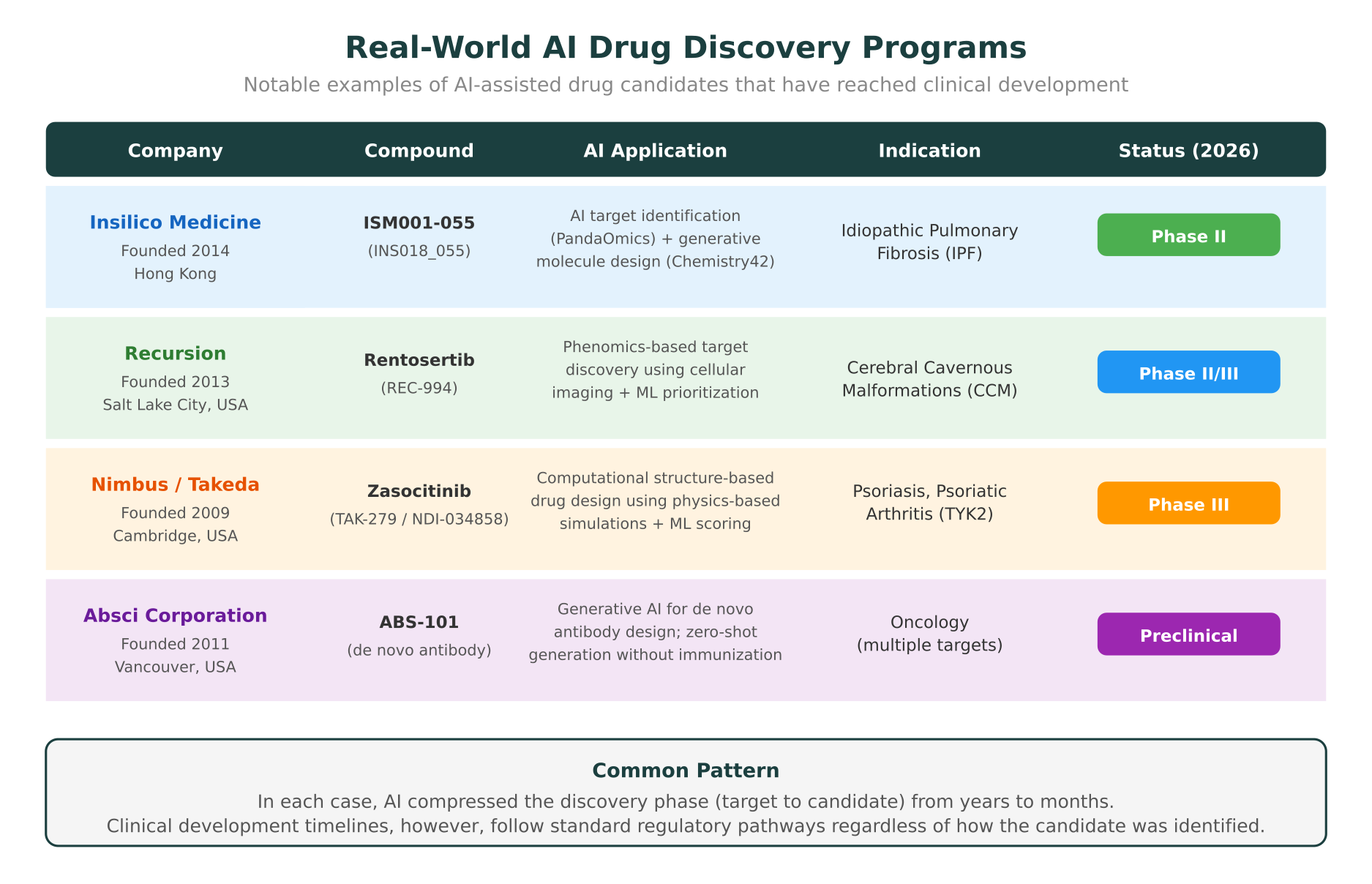
The most convincing evidence for AI’s impact on drug discovery timelines comes from programs that have reached clinical development. As of early 2026, several AI-designed or AI-discovered drug candidates are in human trials.
Insilico Medicine: ISM001-055 (Phase II for IPF)
Insilico Medicine’s ISM001-055 (also known as INS018_055) is arguably the most cited example of end-to-end AI drug discovery. The company used its PandaOmics platform for target identification and Chemistry42 for generative molecule design to discover a novel anti-fibrotic compound for idiopathic pulmonary fibrosis (IPF). The entire process from target identification to preclinical candidate nomination took approximately 18 months, compared to the 4 to 5 years typical for this stage. The compound entered Phase I trials in 2023 and advanced to Phase II in 2024, with data readouts expected in 2026. A paper describing the discovery process was published in Nature Biotechnology in 2024.
Recursion Pharmaceuticals: Rentosertib (Phase II/III for CCM)
Recursion takes a different approach to AI-driven discovery, using high-content cellular imaging and machine learning to identify disease-relevant phenotypes and compounds that reverse them. Their platform screens compounds against cellular models of disease, generating billions of data points that ML models analyze to identify therapeutic candidates. Rentosertib (REC-994), originally identified through this phenomics-based approach, is now in Phase II/III trials for cerebral cavernous malformations (CCM), a rare neurological condition with no approved treatments. Recursion’s platform compressed the target-to-candidate timeline to under two years.
Nimbus Therapeutics: Zasocitinib (Phase III, acquired by Takeda)
Zasocitinib (originally NDI-034858, now TAK-279) is a selective TYK2 inhibitor designed using computational structure-based drug design. Nimbus used physics-based molecular simulations combined with machine learning scoring to design a highly selective allosteric inhibitor of TYK2’s pseudokinase domain, an approach that traditional medicinal chemistry would have struggled to achieve. Takeda acquired the TYK2 program from Nimbus for $6 billion in 2022, validating both the computational approach and the clinical potential. Zasocitinib is now in Phase III trials for psoriasis and psoriatic arthritis.
Absci Corporation: de novo antibody design
Absci has demonstrated generative AI for de novo antibody design, creating therapeutic antibodies from scratch without requiring immunization or library screening. In a study published in Nature Biotechnology in 2024, Absci showed that their generative model could design functional antibodies against HER2 with binding affinities comparable to clinically validated antibodies. While still in preclinical stages, this approach has the potential to compress the antibody discovery phase from 12 to 18 months (traditional hybridoma or phage display) to weeks.
Where AI Is NOT Saving Time
An honest assessment of AI’s impact on drug discovery must acknowledge where current capabilities fall short. The phases that dominate the overall timeline, clinical trials and regulatory review, remain largely resistant to AI-driven acceleration.
Clinical trials: the persistent bottleneck
Clinical trials account for 6 to 10 years of the total drug development timeline. While AI is being applied to trial design (adaptive trial designs, patient stratification, endpoint selection), the fundamental constraints are biological and regulatory, not computational.
Patient recruitment remains the single largest source of delay in clinical trials. Finding patients who meet specific inclusion criteria, obtaining consent, and enrolling them at trial sites is a human-intensive process. AI can improve site selection and patient identification through electronic health record mining, but it cannot accelerate the underlying logistics of recruitment.
Safety monitoring requires observing patients over months or years to detect adverse events. No computational model can substitute for actual observation of drug effects in human biology. Long-term safety data, such as the two-year carcinogenicity studies required for many chronic disease drugs, have irreducible timescales.
Regulatory review operates on its own timeline. The FDA’s standard review cycle is 10 to 12 months for a new molecular entity. Priority review reduces this to 6 to 8 months, but these timescales are set by regulatory process, not by technology.
Manufacturing and scale-up
Process chemistry and manufacturing scale-up remain largely manual disciplines. While AI can suggest synthetic routes, translating a laboratory synthesis to a manufacturing process that reliably produces kilograms of drug substance involves physical optimization that cannot be done in silico. Temperature control, mixing dynamics, crystallization conditions, and purification steps all require empirical optimization at scale.
Biology has irreducible timescales
Certain biological processes simply take time. Toxicology studies require animals to be dosed over specific periods (28-day, 90-day, or chronic studies). Carcinogenicity assessments in rodents require two years. Reproductive toxicity studies follow animals through multiple generations. These timelines are dictated by biology, not by the speed of computation.
The Realistic Picture: What the Numbers Actually Look Like
Combining the phase-by-phase analysis yields a realistic picture of AI’s impact on total drug development timelines.
| Phase | Traditional | AI-Assisted | Time Saved |
|---|---|---|---|
| Target Identification | 1-2 years | 2-6 months | ~75% |
| Hit Finding | 1-2 years | 2-8 weeks | ~85% |
| Lead Optimization | 1-2 years | 6-12 months | ~50% |
| Preclinical | 1-2 years | 1-1.5 years | ~25% |
| Clinical Trials (I-III) | 6-10 years | 5-8 years | ~10-15% |
| Total | 12-15 years | 8-10 years | 3-5 years |
The total time savings of 3 to 5 years is significant. It represents billions of dollars in accelerated revenue for successful drugs and, more importantly, means patients gain access to effective therapies years earlier. But it is not the “drugs in months” narrative that sometimes appears in AI company marketing materials.
The programs that have demonstrated the most dramatic acceleration, like Insilico Medicine’s ISM001-055, compressed the discovery phase (target to IND filing) from 4 to 5 years to approximately 18 months. That is genuinely transformative for early discovery. But ISM001-055 still entered Phase I in 2023 and is expected to complete Phase II in 2026 or 2027, following standard clinical development timelines.
Where the real value compounds
The economic argument for AI in drug discovery is actually stronger than the timeline argument alone. Three factors compound the value beyond simple time savings:
-
Higher success rates through better target selection. Programs with genetic evidence supporting the target are roughly twice as likely to succeed in clinical trials, as documented by AstraZeneca and others. AI-powered computational drug target discovery systematically identifies targets with stronger evidence bases, potentially improving the 90% clinical failure rate.
-
Lower cost per program. Fewer physical screening campaigns, fewer synthesis cycles, and fewer failed preclinical candidates all reduce the cost of the discovery phase. Even if clinical trial costs remain unchanged, reducing discovery costs from $500 million to $100 million per successful drug is economically significant.
-
More shots on goal. By compressing and cheapening the discovery phase, AI enables organizations to pursue more programs simultaneously. A portfolio of 10 AI-enabled programs may cost less than 3 traditional programs, dramatically improving the odds that at least one succeeds.
How Molecular Intelligence Platforms Fit In
The practical challenge for research teams is not the availability of AI tools. It is the fragmentation of those tools across dozens of disconnected platforms, databases, and interfaces. A typical AI-assisted drug discovery workflow might involve Open Targets for target identification, AlphaFold for structure prediction, DiffDock for virtual screening, separate ADMET prediction models, and manual querying of ClinVar, gnomAD, UniProt, and PDB for supporting evidence.
Molecular intelligence platforms address this by integrating multiple capabilities into a single workspace. For teams working on early-stage drug discovery, the ability to query biological databases through natural language, run structural analysis including protein stability predictions, and access 30+ clinical and research databases from one interface compresses the “evidence gathering” phase of target identification from weeks to hours.
For a broader perspective on how AI is transforming biological research beyond drug discovery, including protein design, clinical genomics, and proteomics analysis, see our overview of what is actually working in AI for biology in 2026.
What Comes Next
Several trends will shape AI’s impact on drug discovery timelines over the next two to three years.
Foundation models for biology. Large-scale models trained on multi-omics data are enabling more sophisticated reasoning across data types. These models can identify target-disease connections that no single data source would reveal, further compressing the target identification phase.
Closed-loop autonomous labs. Companies like Recursion and Arctoris are combining AI-guided experiment design with robotic automation to create semi-autonomous DMTA cycles. When the AI can design an experiment, a robot executes it, and the results feed directly back into the model, cycle times drop from weeks to days.
Improved clinical trial design. While AI cannot eliminate the biological timescales of clinical trials, it can improve trial efficiency. Adaptive trial designs that use Bayesian modeling to adjust dosing, enrollment, and endpoints during the trial can reduce the number of patients needed and the overall trial duration by 20 to 30%, according to analyses published in Clinical Pharmacology and Therapeutics in 2024.
Regulatory adaptation. Regulatory agencies are beginning to develop frameworks for evaluating AI-designed drugs. The FDA’s discussion papers on AI in drug development, published in 2024 and 2025, signal a willingness to adapt review processes as the evidence base for AI-assisted discovery matures.
The honest conclusion is that AI is delivering real, measurable compression of drug discovery timelines, primarily in the early stages. The total impact, reducing development from 12 to 15 years to perhaps 8 to 10 years, is significant but not revolutionary. The revolution, if it comes, will require AI to also address the clinical trial bottleneck, which is as much a regulatory and biological challenge as a computational one.
Researchers interested in exploring AI-assisted drug discovery workflows can apply for up to $10,000 in free MIP credits to run multi-omics analyses, structural assessments, and database queries on Purna’s platform.
Purna AI’s Molecular Intelligence Platform (MIP) is an AI-powered workspace for biology teams. It brings together molecular analysis, variant interpretation, protein structure prediction, and clinical database integrations into one environment. Built for teams who work with biological data and need consistent, reproducible answers without juggling disconnected tools. Learn more at purna.ai.
Explore Purna's Molecular Intelligence Platform
AI-powered workspace for biology teams to accelerate drug discovery from target identification to lead optimization.
Try Purna AI →
