Computational Drug Target Discovery: A Guide for Research Teams

Most drug programs fail not because the molecule is wrong, but because the target is. Roughly 90% of drug candidates entering clinical trials never reach approval, and analyses by AstraZeneca and other large pharmaceutical companies have consistently identified poor target selection as the single largest contributor to clinical attrition. A study published in Nature Reviews Drug Discovery in 2022 found that drug programs with genetic evidence supporting the target-disease link are approximately twice as likely to succeed in clinical development.
This creates a clear imperative: the more evidence a team can assemble for a candidate target before committing to a drug program, the higher the probability of clinical success. Computational target discovery has matured significantly over the past five years, driven by the availability of large-scale genomic datasets, AI-predicted protein structures, and integrative platforms that combine multiple evidence types. This guide covers the key computational approaches for identifying and validating drug targets, the databases and tools that support each stage, and practical considerations for building a target discovery workflow.
The Target Discovery Pipeline: From Data to Decision
Drug target discovery is not a single step. It is a multi-stage process that progressively narrows a large space of potential targets down to a small set of validated, druggable candidates. Each stage generates different types of evidence, and the strongest target selections are those supported by convergent evidence across multiple stages.
The four primary stages are:
- Target identification using multi-omics data (genomics, transcriptomics, proteomics, metabolomics)
- Genetic validation using causal inference methods (Mendelian randomization, loss-of-function variants, PheWAS)
- Structural druggability assessment using binding site prediction and protein structure analysis
- AI-based prioritization using knowledge graphs, network analysis, and literature mining
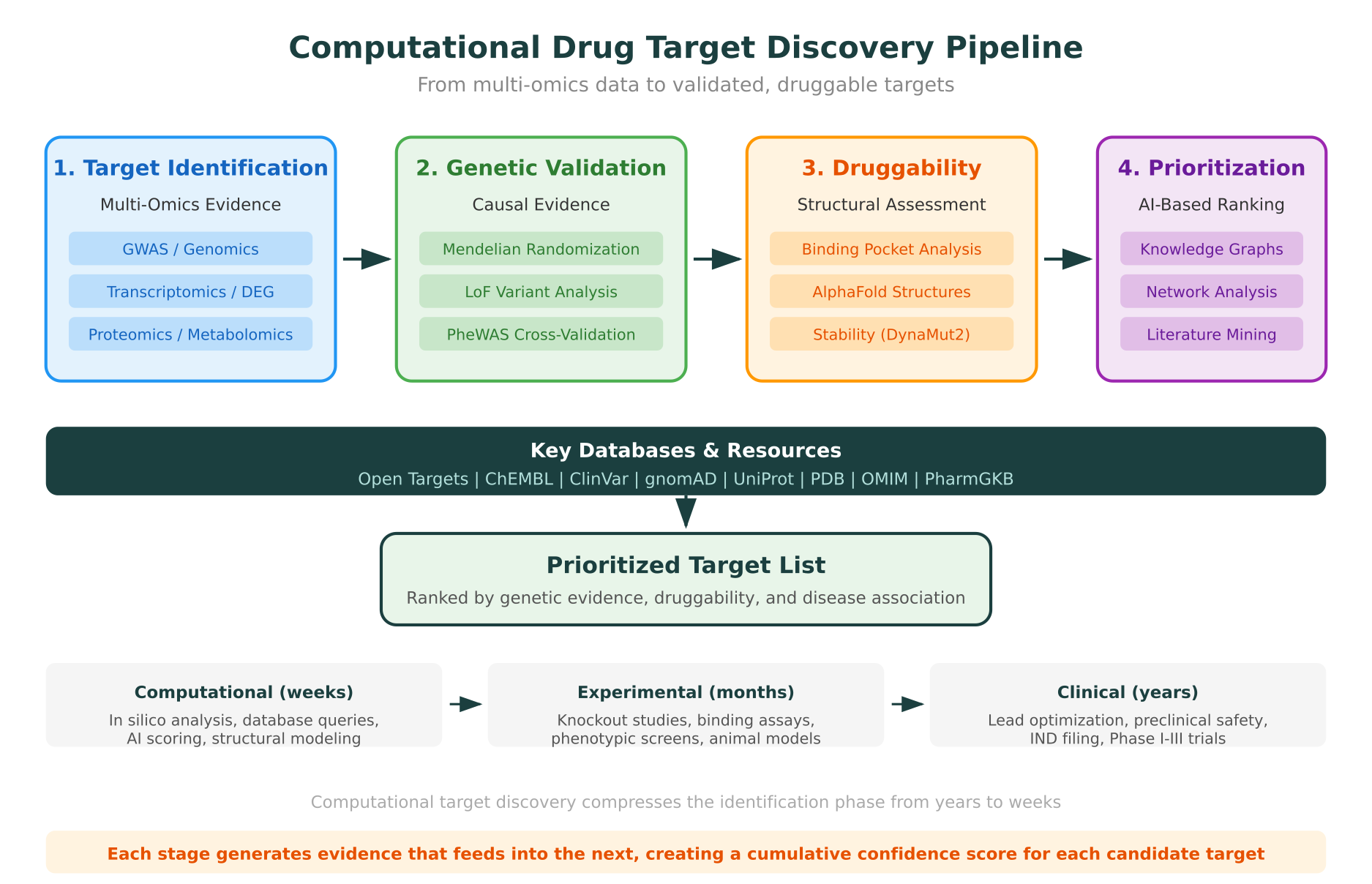
Teams that work across all four stages, rather than relying on any single evidence type, consistently make better target selections. The remainder of this guide covers each stage in detail.
Multi-Omics Target Identification
The starting point for most target discovery programs is identifying genes or proteins that are differentially active in disease. Modern computational approaches draw on multiple omics layers simultaneously, because no single data type provides a complete picture.
Genomics: GWAS as the foundation
Genome-wide association studies (GWAS) have identified over 350,000 trait-associated genetic loci as of early 2026. For drug target discovery, the key insight from GWAS is not the individual SNP, but the gene it implicates. Locus-to-gene mapping, which connects a significant GWAS signal to the most likely causal gene, remains one of the most active areas of computational genetics.
The Open Targets Platform, a joint initiative between the Wellcome Sanger Institute, EMBL-EBI, and several pharmaceutical companies, provides systematic target-disease association scores that integrate GWAS data with functional genomics, expression data, and clinical evidence. Each target receives a composite score between 0 and 1 for its association with a given disease, broken down by evidence type. This scoring framework gives research teams a structured starting point rather than requiring them to manually curate evidence from dozens of sources.
The GWAS Catalog, maintained by EMBL-EBI and NHGRI, provides the underlying curated dataset of published GWAS findings. As of early 2026, it contains over 600,000 variant-trait associations from more than 7,000 publications. For teams working on target identification, the GWAS Catalog serves as the primary source of genetic association evidence.
Transcriptomics: differential expression signatures
Transcriptomic profiling, typically via RNA-seq, identifies genes whose expression levels differ significantly between disease and healthy tissue. Differential gene expression (DGE) analysis highlights potential targets that are upregulated in disease (candidates for inhibition) or downregulated (candidates for activation or replacement therapy).
Large-scale resources like the Genotype-Tissue Expression (GTEx) project and the Cancer Genome Atlas (TCGA) provide reference datasets for normal and disease tissue expression. Single-cell RNA sequencing has added resolution, revealing cell-type-specific expression patterns that bulk RNA-seq misses. A target that appears upregulated in bulk tissue analysis may actually be expressed only in a specific immune cell subset, which has implications for both the therapeutic strategy and potential side effects.
For teams working across genomics, proteomics, and transcriptomics, integrating these data types is essential for building a complete picture of target biology.
Proteomics and metabolomics
Proteomic profiling provides a more direct readout of protein abundance and post-translational modifications. Mass spectrometry-based proteomics, particularly data-independent acquisition (DIA) workflows, now routinely quantify thousands of proteins in clinical samples. Proximity extension assays (Olink) and aptamer-based platforms (SomaScan) enable high-throughput plasma proteomics, which is particularly useful for identifying secreted or circulating targets.
Metabolomics complements the picture by measuring downstream functional outputs, small molecules whose levels change in disease states. Metabolomic signatures can point to pathway disruptions that are not visible at the gene or protein level, sometimes implicating enzymes or transporters as drug targets.
The power of multi-omics target identification lies in convergence. A gene that shows both a significant GWAS signal and differential expression in disease tissue, with corresponding changes at the protein and metabolite level, is a much stronger candidate than one supported by a single data type.
Genetic Evidence for Target Validation
Identifying a target is only the first step. Validating that modulating the target will have the desired therapeutic effect, without unacceptable toxicity, is where most programs succeed or fail. Genetic evidence has emerged as the most reliable predictor of clinical success.
Mendelian randomization
Mendelian randomization (MR) uses naturally occurring genetic variants as instruments to estimate the causal effect of a target on a disease outcome. The logic is elegant: if genetic variants that reduce the activity of a protein are associated with lower disease risk, then a drug that inhibits that protein should, in principle, have the same effect.
Large-scale MR studies using data from the UK Biobank (approximately 500,000 participants) and FinnGen (approximately 500,000 participants) have become standard in target validation. A landmark analysis published in Nature Genetics in 2021 by Zheng et al. systematically applied MR to evaluate drug targets across hundreds of diseases, identifying both novel target-disease connections and potential safety signals.
Two-sample MR, which uses summary statistics from separate exposure and outcome GWAS, has made this approach accessible to teams without access to individual-level cohort data. Colocalization analysis (tools like coloc and COLOC2) adds an important check by verifying that the GWAS signal for the target and the GWAS signal for the disease are driven by the same causal variant, not two different variants in linkage disequilibrium.
Loss-of-function variants as natural experiments
Loss-of-function (LoF) variants in human populations serve as natural knockouts. The gnomAD database, now containing exome and genome data from over 800,000 individuals, catalogs the frequency of protein-truncating variants across human genes. Genes that are highly intolerant to LoF variation (low pLI or high LOEUF score) are likely essential, which carries implications for both target validation and safety assessment.
The converse is equally informative. Individuals who carry LoF variants in a candidate target without apparent health consequences provide evidence that the target can be safely inhibited. The most famous example is PCSK9: the discovery that individuals with naturally occurring LoF variants in PCSK9 had substantially lower LDL cholesterol and reduced cardiovascular risk directly supported the development of PCSK9 inhibitors, which are now approved therapies.
Phenome-wide association studies (PheWAS)
PheWAS inverts the GWAS approach: instead of testing many variants against one phenotype, it tests variants in a candidate target gene against thousands of phenotypes. This reveals pleiotropic effects, cases where modulating a target affects multiple organ systems or disease outcomes.
For target validation, PheWAS is primarily a safety tool. It identifies potential on-target side effects before a drug program enters preclinical development. If variants in a candidate target gene are associated with increased risk of liver disease, cardiac arrhythmia, or immune suppression, those findings should inform the risk-benefit assessment early in the program.
The PheWAS approach has been applied at scale using electronic health record data from biobanks including the UK Biobank, the Million Veteran Program, and the All of Us Research Program. Tools like PheWAS and PHESANT automate the statistical analysis across thousands of phenotypes.
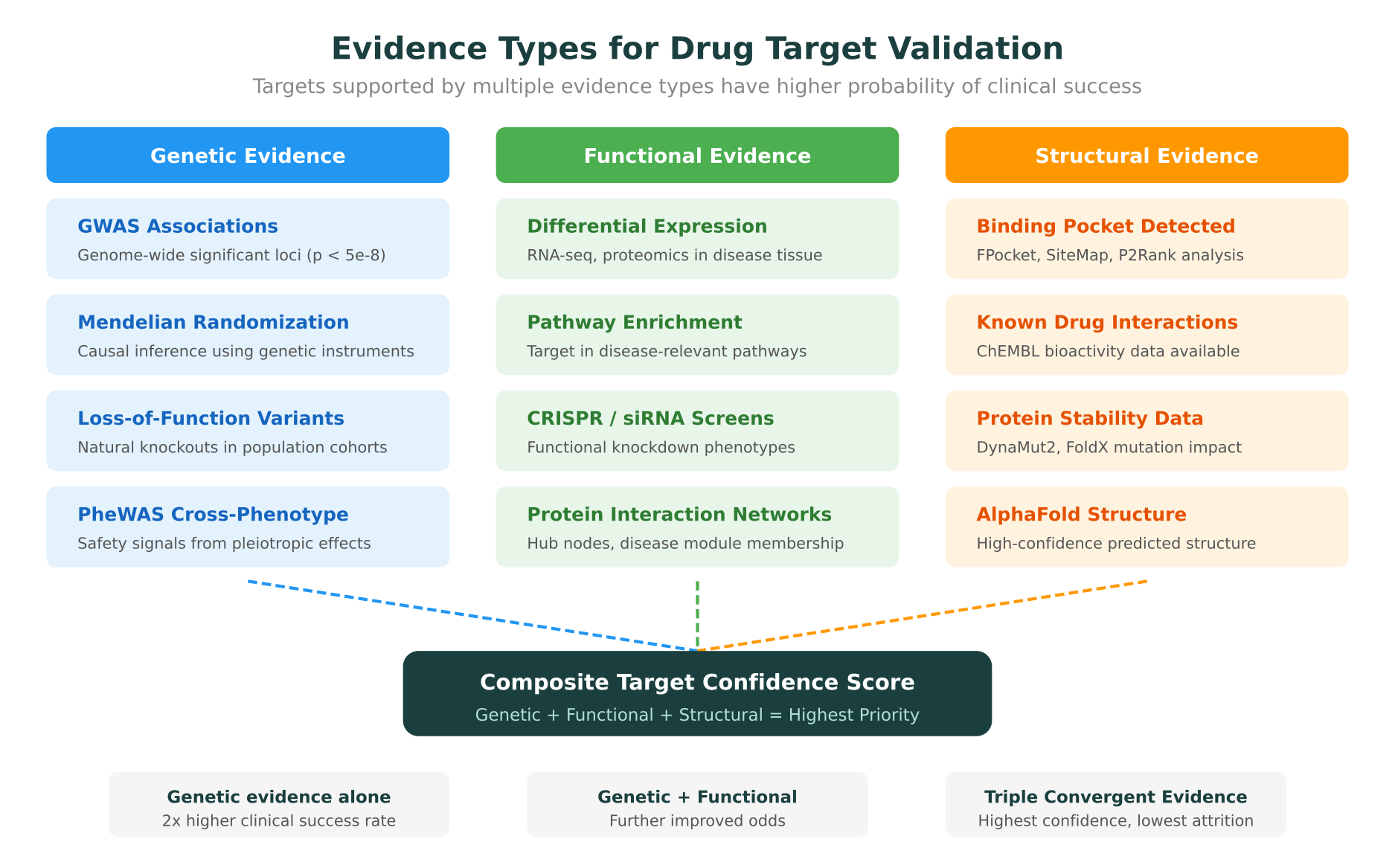
Structural Druggability Assessment
A biologically validated target is only useful if it can be modulated by a drug molecule. Structural druggability assessment evaluates whether a protein has binding sites that can accommodate small molecules, peptides, or biologics.
Binding site prediction
Computational pocket detection tools identify cavities on the protein surface that are large enough, deep enough, and appropriately shaped to bind drug-like molecules. Established tools include:
- FPocket (open-source), which uses Voronoi tessellation and alpha spheres to detect pockets. It ranks cavities by a druggability score based on physicochemical properties.
- SiteMap (Schrodinger), which evaluates pockets based on size, enclosure, hydrophobicity, hydrogen bonding potential, and exposure to solvent.
- P2Rank (open-source, machine learning-based), which uses random forests trained on features extracted from the protein surface to predict binding sites. Published in Journal of Cheminformatics in 2018, P2Rank has shown strong performance on diverse benchmark sets.
- DeepSite and DeepPocket, neural network-based methods that learn pocket features directly from 3D protein structures.
A target with a large, enclosed, hydrophobic pocket is a strong small-molecule candidate. Shallow or highly charged surfaces are more challenging for small molecules but may be addressable with peptides, PROTACs, or molecular glues.
AlphaFold structures for novel targets
Before AlphaFold, structural druggability assessment was limited to proteins with experimental crystal structures or cryo-EM structures in the PDB. The AlphaFold Protein Structure Database, which now contains predicted structures for over 200 million proteins, has dramatically expanded the number of targets that can be assessed computationally.
However, using AlphaFold structures for druggability analysis requires care. The confidence metric (pLDDT) indicates how reliable each region of the predicted structure is. Binding site predictions on low-confidence regions (pLDDT below 70) should be treated cautiously. Additionally, AlphaFold predicts a single static conformation, while many binding sites are only accessible in specific conformational states. Teams comparing structure prediction tools should consider the strengths of different approaches, as discussed in our comparison of AlphaFold 3, Boltz-2, and ESMFold.
For targets where experimental structures are unavailable, a practical workflow combines AlphaFold structure prediction, pocket detection with FPocket or P2Rank, and stability analysis using tools like DynaMut2 to assess how mutations in the binding region affect protein stability. Understanding how mutations impact protein structure and function is directly relevant to evaluating whether a target’s binding site is robust enough for drug development.
Assessing druggability beyond small molecules
The druggability landscape has expanded considerably beyond traditional small-molecule binding pockets. Protein degradation approaches (PROTACs, molecular glues) can target proteins that lack conventional binding sites by recruiting E3 ubiquitin ligases. Allosteric modulators target sites distant from the active site. Antisense oligonucleotides and siRNA bypass the protein entirely by targeting mRNA.
This means that a protein deemed “undruggable” by traditional pocket analysis may still be targetable. Computational tools are adapting: CryptoSite predicts hidden allosteric sites that are only transiently accessible, and protein-protein interaction interface analysis tools like TIMBAL identify hotspots at protein interfaces that can be targeted by peptides or stapled helices.
AI-Based Target Prioritization
After assembling multi-omics evidence, genetic validation data, and structural druggability assessments for a set of candidate targets, research teams face a prioritization challenge. AI-based approaches help rank targets by integrating heterogeneous evidence types and identifying patterns that manual analysis might miss.
Knowledge graphs for target discovery
Knowledge graphs represent biological entities (genes, proteins, diseases, drugs, pathways) and their relationships as a network. Graph-based reasoning then identifies connections that suggest novel target-disease relationships or drug repurposing opportunities.
Several large-scale biomedical knowledge graphs are available:
- DRKG (Drug Repurposing Knowledge Graph), built by Amazon, contains over 97,000 entities and 5.9 million relationships drawn from DrugBank, Hetionet, GNBR, STRING, and IntAct.
- Hetionet, published in eLife in 2017, integrates 47,031 nodes of 11 types and 2.25 million edges of 24 types, connecting compounds, diseases, genes, and anatomical structures.
- BioKG and PrimeKG provide updated, comprehensive knowledge graphs that include protein structure features and pathway annotations.
Link prediction on these graphs, using graph neural networks or knowledge graph embedding methods like TransE, RotatE, and ComplEx, can identify high-confidence target-disease associations that are not yet established in the literature. A study by Bonner et al. published in Nature Machine Intelligence in 2022 demonstrated that knowledge graph embeddings could predict drug-target interactions with strong performance on held-out test sets.
Network-based approaches
Protein-protein interaction (PPI) networks provide a different lens on target prioritization. Tools like STRING (which integrates known and predicted protein interactions) and BioGRID (which catalogs experimentally validated interactions) map the interaction landscape around candidate targets.
Network metrics matter for target selection. Targets that occupy hub positions in disease-relevant network modules are more likely to have significant biological effects when modulated. However, highly connected hub proteins may also have more off-target effects. Network-based approaches like diffusion algorithms and random walk with restart can propagate disease association signals through the network, identifying genes that are functionally close to known disease genes even if they lack direct genetic evidence.
Literature mining and NLP
The biomedical literature contains vast amounts of target-relevant information that is difficult to access manually. Over 36 million articles in PubMed, plus preprints, patents, and clinical trial records, create an information landscape that no individual researcher can survey comprehensively.
Biomedical NLP tools have improved substantially. PubMedBERT and BioGPT, transformer-based language models trained on biomedical text, power named entity recognition (NER) systems that extract gene-disease relationships, drug-target interactions, and mechanism-of-action statements from published literature. Tools like PubTator 3 and SciBERT provide pre-trained models that can be fine-tuned for specific extraction tasks.
For research teams, the practical value of literature mining is in surfacing connections that might otherwise be overlooked: a case report describing an unexpected phenotype in patients taking a drug that inhibits a candidate target, or a pre-clinical study in a related species that provides functional evidence for the target.
The ability to query biological databases using natural language is particularly valuable in this context, because it allows researchers to synthesize evidence from multiple sources without writing custom queries for each database.
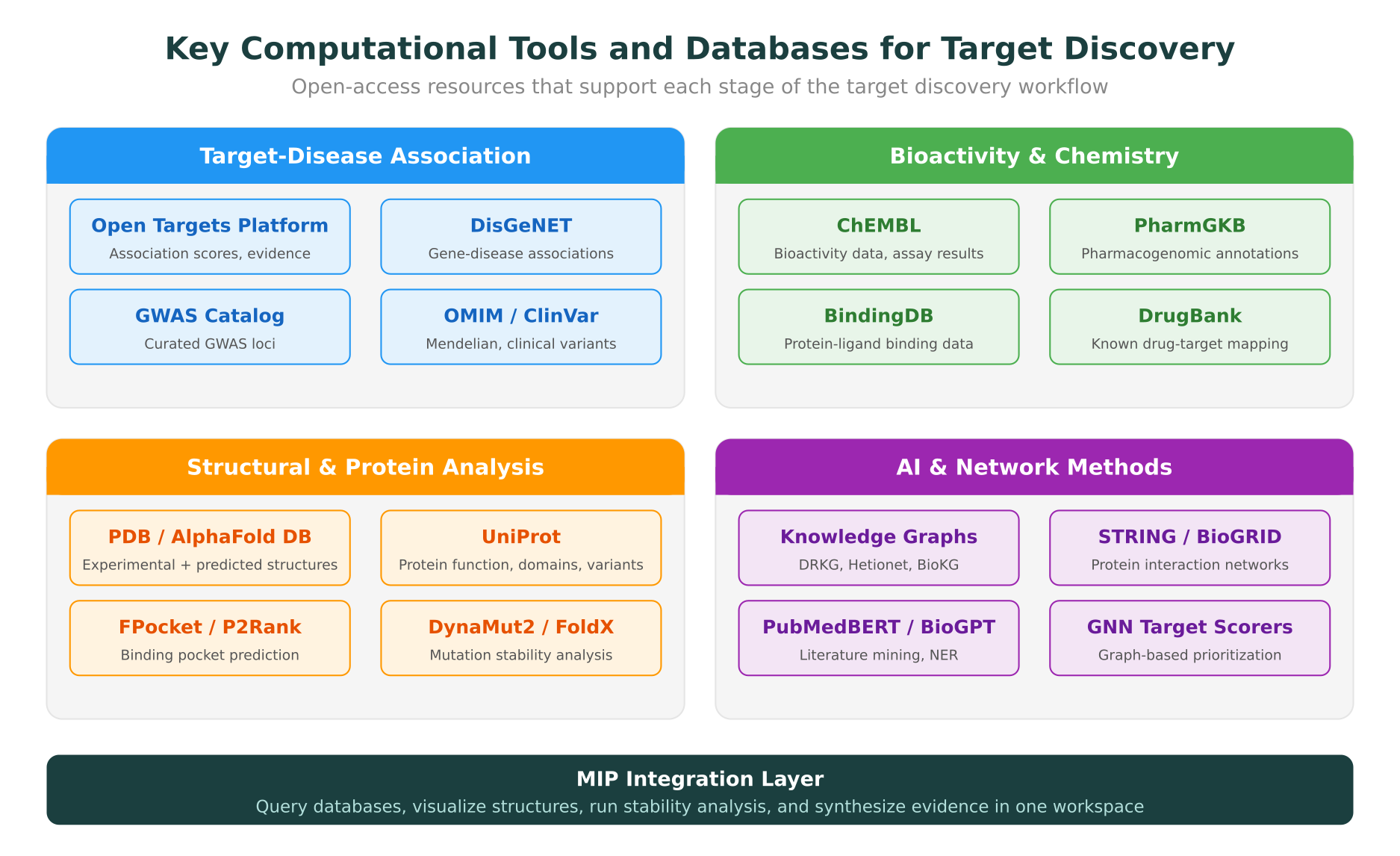
Key Databases and Resources for Target Discovery
Building an effective target discovery workflow requires familiarity with the major databases and platforms. The table below summarizes the most important resources for each stage of the pipeline.
| Resource | Type | Primary Use | Access |
|---|---|---|---|
| Open Targets Platform | Target-disease associations | Composite association scores from multiple evidence types | Open |
| ChEMBL | Bioactivity database | Small-molecule activity data against protein targets | Open |
| GWAS Catalog | Genetic associations | Curated GWAS significant associations | Open |
| gnomAD | Population genetics | LoF variant frequencies, constraint metrics | Open |
| ClinVar | Clinical variants | Pathogenic/benign variant classifications | Open |
| OMIM | Mendelian genetics | Gene-disease relationships for monogenic disorders | Open |
| UniProt | Protein knowledge base | Protein function, domains, variants, annotations | Open |
| PDB | Structural biology | Experimental protein structures | Open |
| AlphaFold DB | Predicted structures | AI-predicted protein structures (200M+ proteins) | Open |
| PharmGKB | Pharmacogenomics | Drug-gene-phenotype relationships | Open |
| STRING | PPI network | Protein-protein interaction scores | Open |
| DisGeNET | Gene-disease | Aggregated gene-disease associations | Open (basic) |
| DrugBank | Drug knowledge base | Drug-target mapping, mechanism of action | Free (academic) |
| BindingDB | Binding data | Measured protein-ligand binding affinities | Open |
The challenge for most research teams is not access to individual databases, which are predominantly open, but the overhead of querying multiple databases, reconciling identifiers, and synthesizing evidence into a coherent assessment for each candidate target.
Target-Disease Association Scoring
Quantitative scoring frameworks help standardize how teams evaluate and compare candidate targets. The Open Targets Platform provides the most widely used scoring system, assigning each target-disease pair an overall association score based on weighted contributions from seven evidence categories: genetic associations, somatic mutations, known drugs, pathways, RNA expression, literature mining, and animal models.
Beyond Open Targets, teams often build custom scoring frameworks tailored to their therapeutic area. A typical composite score might weight:
- Genetic evidence (GWAS, MR, LoF): 30-40% of total weight, given its demonstrated correlation with clinical success
- Expression evidence (differential expression, tissue specificity): 15-20%
- Structural druggability (pocket druggability score, structure availability): 15-20%
- Existing pharmacology (known modulators, selectivity data): 10-15%
- Safety signals (PheWAS, essential gene constraint, expression in safety-relevant tissues): 10-15%
The specific weights should reflect the team’s priorities and the therapeutic area. Oncology programs may tolerate higher safety risk for essential gene targets. Chronic disease programs require a higher safety bar. The key principle is that the scoring framework makes the decision criteria explicit and reproducible, rather than relying on implicit expert judgment that varies across team members.
How MIP Supports Target Discovery Workflows
The target discovery workflow described above requires researchers to move between multiple databases, visualization tools, and analysis platforms. Purna AI’s Molecular Intelligence Platform (MIP) provides an integrated environment that reduces this fragmentation.
Structural analysis in one workspace
MIP pulls protein structures from the PDB or AlphaFold and renders them in Molstar for interactive 3D visualization. Researchers can inspect predicted binding pockets, examine residue-level contacts, and overlay mutations of interest directly in the browser. For druggability assessment, DynaMut2 integration enables rapid estimation of how mutations in or near binding sites affect protein stability (delta-delta-G calculations), which is relevant both for understanding disease-associated variants and for evaluating the robustness of a potential drug binding site.
Database integration across 30+ sources
Rather than querying UniProt, PDB, OMIM, ClinVar, gnomAD, and PharmGKB separately, MIP connects to over 30 clinical and biological databases through a unified interface. This is particularly valuable during the evidence synthesis phase of target discovery, when a researcher needs to rapidly compile genetic evidence, functional annotations, known pharmacology, and structural data for a set of candidate targets. Every answer includes citations traceable to the source database, supporting reproducibility and audit trails.
Natural language queries
For researchers who are not computational biologists, MIP’s natural language interface makes it possible to query target evidence without writing database-specific queries or code. Questions like “What is the genetic evidence linking PCSK9 to cardiovascular disease?” or “Show me the binding pocket of BRAF V600E” translate into structured queries across the relevant databases and return synthesized answers with references. This capability is discussed in detail in our guide on querying biological databases with natural language.
Bioinformatics code execution
For teams that do have computational expertise, MIP provides containerized bioinformatics environments where researchers can write and execute custom analyses, whether that involves running a Mendelian randomization analysis in R, performing network analysis with NetworkX, or building a custom target scoring pipeline. Having the analysis environment co-located with the database access and structural tools eliminates the overhead of moving data between disconnected systems.
Practical Recommendations for Research Teams
Building a target discovery workflow from scratch is a substantial undertaking. Based on published best practices and the experience of computational biology teams at pharmaceutical companies and academic research groups, several recommendations are worth highlighting:
-
Start with genetic evidence. The correlation between genetic support and clinical success is the most robust finding in target validation. Prioritize candidates with GWAS support, and use MR to strengthen the causal case.
-
Integrate multiple evidence types. A target supported by genetic, transcriptomic, and structural evidence is a fundamentally stronger candidate than one supported by any single data type. Build scoring frameworks that reward convergent evidence.
-
Assess druggability early. A biologically compelling target that cannot be modulated by any available drug modality is not a viable starting point for a drug program. Run pocket analysis on AlphaFold structures as soon as a target enters your pipeline.
-
Use PheWAS for safety de-risking. Pleiotropic effects identified through PheWAS can surface safety concerns before preclinical investment. This is particularly important for chronic disease targets where the therapeutic window may be narrow.
-
Leverage existing scoring frameworks. Open Targets provides a well-validated, transparent scoring system. Use it as a baseline and customize weights for your therapeutic area rather than building a scoring system from scratch.
-
Automate evidence collection. Manually querying multiple databases for each candidate target is slow and error-prone. Platforms that integrate multiple data sources, whether through APIs, unified interfaces, or natural language queries, reduce the time from candidate identification to evidence-based prioritization.
-
Document and version your evidence. Target selection decisions have downstream consequences that span years. Ensure that the evidence base, scoring framework, and rationale are documented in a way that can be audited and revisited as new data emerges.
For teams exploring how molecular intelligence is reshaping drug discovery, computational target discovery represents one of the areas where the impact is most immediate and measurable.
Looking Ahead
The next phase of computational target discovery will be shaped by several trends. Foundation models trained on multi-omics data are enabling more sophisticated integration of genetic, expression, and structural evidence. Large-scale biobank data from programs like the UK Biobank, All of Us, and FinnGen continue to expand the genetic evidence base. And the combination of AI-predicted structures with increasingly accurate pocket detection and molecular docking tools is making structural druggability assessment more reliable for novel targets.
For a broader perspective on how AI is driving change across biology, including drug discovery, protein design, and clinical genomics, see our overview of what is actually working in AI for biology in 2026.
Researchers interested in exploring computational target discovery workflows can apply for up to $10,000 in free MIP credits to run multi-omics analyses, structural assessments, and database queries on Purna’s platform.
Purna AI’s Molecular Intelligence Platform (MIP) is an AI-powered workspace for biology teams. It brings together molecular analysis, variant interpretation, protein structure prediction, and clinical database integrations into one environment. Built for teams who work with biological data and need consistent, reproducible answers without juggling disconnected tools. Learn more at purna.ai.
Explore Purna's Molecular Intelligence Platform
AI-powered workspace for biology teams to accelerate drug discovery from target identification to lead optimization.
Try Purna AI →
