AI in Proteomics: From Mass Spectrometry to Protein Function Prediction

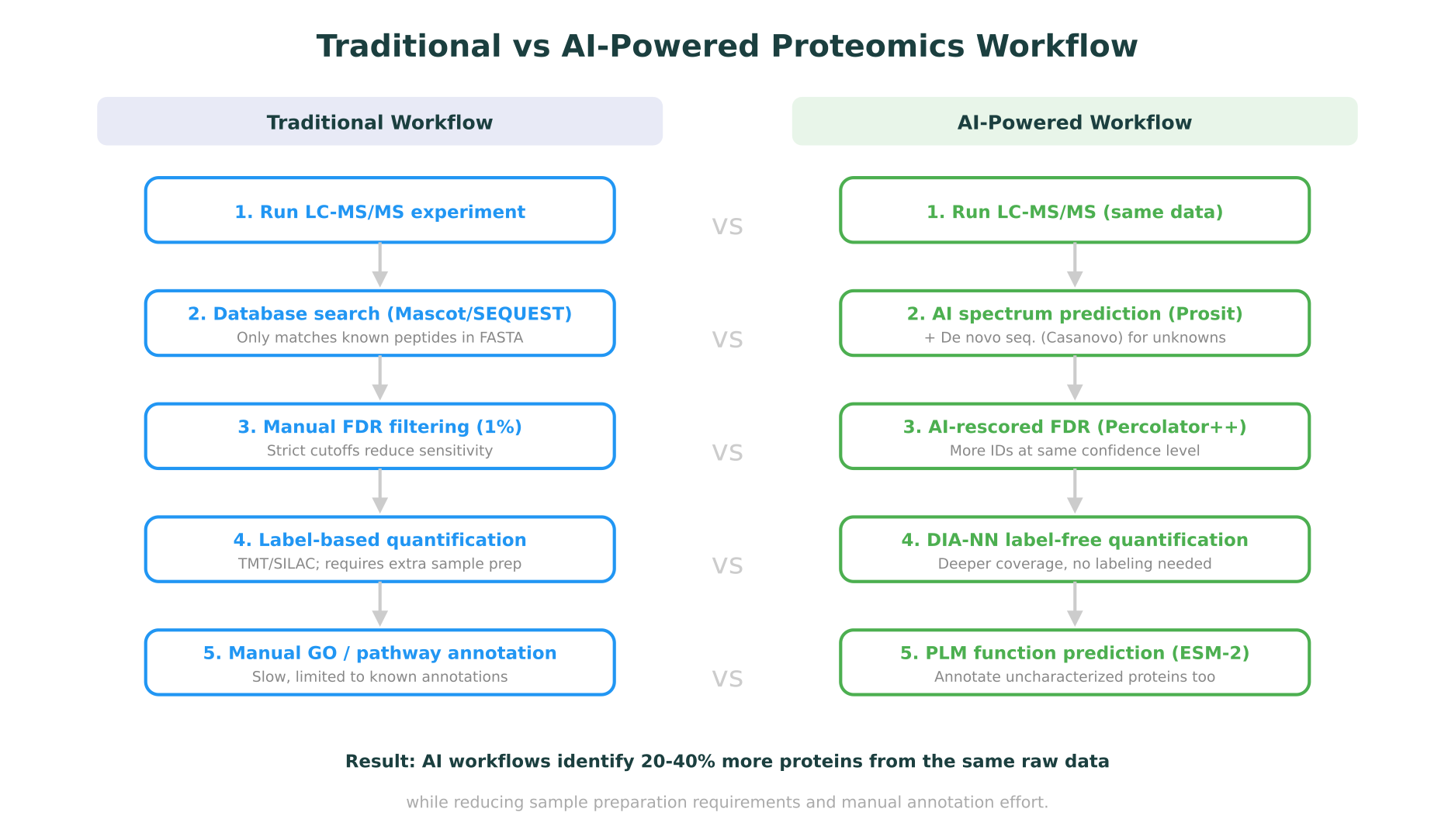
Proteomics has always been a data-intensive field. A single liquid chromatography-tandem mass spectrometry (LC-MS/MS) experiment can generate hundreds of thousands of spectra, each representing a fragmented peptide that needs to be identified, quantified, and mapped back to its parent protein. For decades, the computational bottleneck in proteomics was not generating data but interpreting it. Traditional database search engines like Mascot and SEQUEST matched experimental spectra against theoretical fragmentation patterns derived from protein sequence databases, a process that was effective but inherently limited to known sequences and predefined search spaces.
AI is changing this. Over the past three years, deep learning models have reshaped nearly every step of the proteomics pipeline: spectrum prediction, peptide identification, de novo sequencing, protein quantification, post-translational modification (PTM) discovery, and protein function annotation. These are not incremental improvements. In several cases, AI-powered tools have expanded the detectable proteome by 20-40% from the same raw data, identified novel peptides invisible to traditional methods, and predicted protein functions for sequences with no experimental annotation.
This post maps the current landscape of AI in proteomics, covering the tools, methods, and results that matter for computational biologists and proteomics researchers in 2026. It connects proteomics-specific advances to the broader context of multi-omics integration and shows where platforms built around molecular intelligence are making these capabilities accessible beyond specialized bioinformatics teams.
AI-Powered Peptide Identification
The foundation of any proteomics experiment is peptide identification: determining which peptides are present in a sample based on their fragmentation spectra. Traditional approaches rely on comparing experimental MS/MS spectra against theoretical spectra generated from protein sequence databases. This works well for organisms with complete reference proteomes, but struggles with novel peptides, unexpected modifications, and organisms with incomplete databases.
Spectrum prediction with Prosit and MS2PIP
Rather than generating theoretical spectra from simple fragmentation rules, AI models learn the complex, context-dependent patterns of peptide fragmentation directly from large experimental datasets.
Prosit, developed at the Technical University of Munich and published in Nature Methods (2019), was one of the first deep learning models to predict peptide fragmentation spectra and retention times with high fidelity. Trained on millions of high-quality spectra from the ProteomeTools project, Prosit uses a recurrent neural network to predict fragment ion intensities for any given peptide sequence and charge state. The predicted spectra are close enough to experimental reality that they can replace experimental spectral libraries in data-independent acquisition (DIA) analysis, eliminating the need to generate sample-specific libraries.
MS2PIP takes a complementary approach, using gradient-boosted trees (and more recently, transformer architectures) to predict fragment ion intensities from peptide sequences. Published in Nucleic Acids Research (2019) and continuously updated, MS2PIP provides fast spectrum prediction that integrates directly into rescoring pipelines.
DeepLC focuses specifically on retention time prediction, using a convolutional neural network to predict when a peptide will elute from the chromatographic column. Accurate retention time predictions serve as an additional scoring dimension during peptide identification, reducing false positives. DeepLC was published in Nature Methods in 2021 and has become a standard component in modern proteomics pipelines.
The practical impact of these tools is significant. When Prosit-predicted spectra are used to rescore peptide-spectrum matches identified by traditional search engines, the number of confidently identified peptides at a 1% false discovery rate (FDR) increases substantially. A 2023 benchmarking study in Molecular and Cellular Proteomics showed that AI-based rescoring improved peptide identifications by 20-30% compared to traditional target-decoy approaches alone.
De novo sequencing with Casanovo
Database search methods are inherently limited: they can only identify peptides present in the reference database. For immunopeptidomics, metaproteomics, antibody sequencing, and any application involving non-canonical peptides, de novo sequencing (determining the peptide sequence directly from the spectrum without a database) is essential.
Casanovo, published in Nature Machine Intelligence in 2024, represents the current state of the art in AI-driven de novo peptide sequencing. Built on a transformer architecture, Casanovo treats spectrum-to-sequence prediction as a translation problem: the input is a mass spectrum, and the output is a peptide sequence, one amino acid at a time. Trained on over 30 million peptide-spectrum matches from diverse organisms and instrument types, Casanovo achieves amino acid-level recall rates above 70% on held-out datasets, a meaningful improvement over earlier methods like DeepNovo and PointNovo.
What makes Casanovo particularly useful is its ability to identify peptides that would be missed entirely by database search. In immunopeptidomics applications, where MHC-presented peptides often include non-tryptic cleavages and post-translational modifications absent from standard databases, de novo sequencing fills a critical gap.
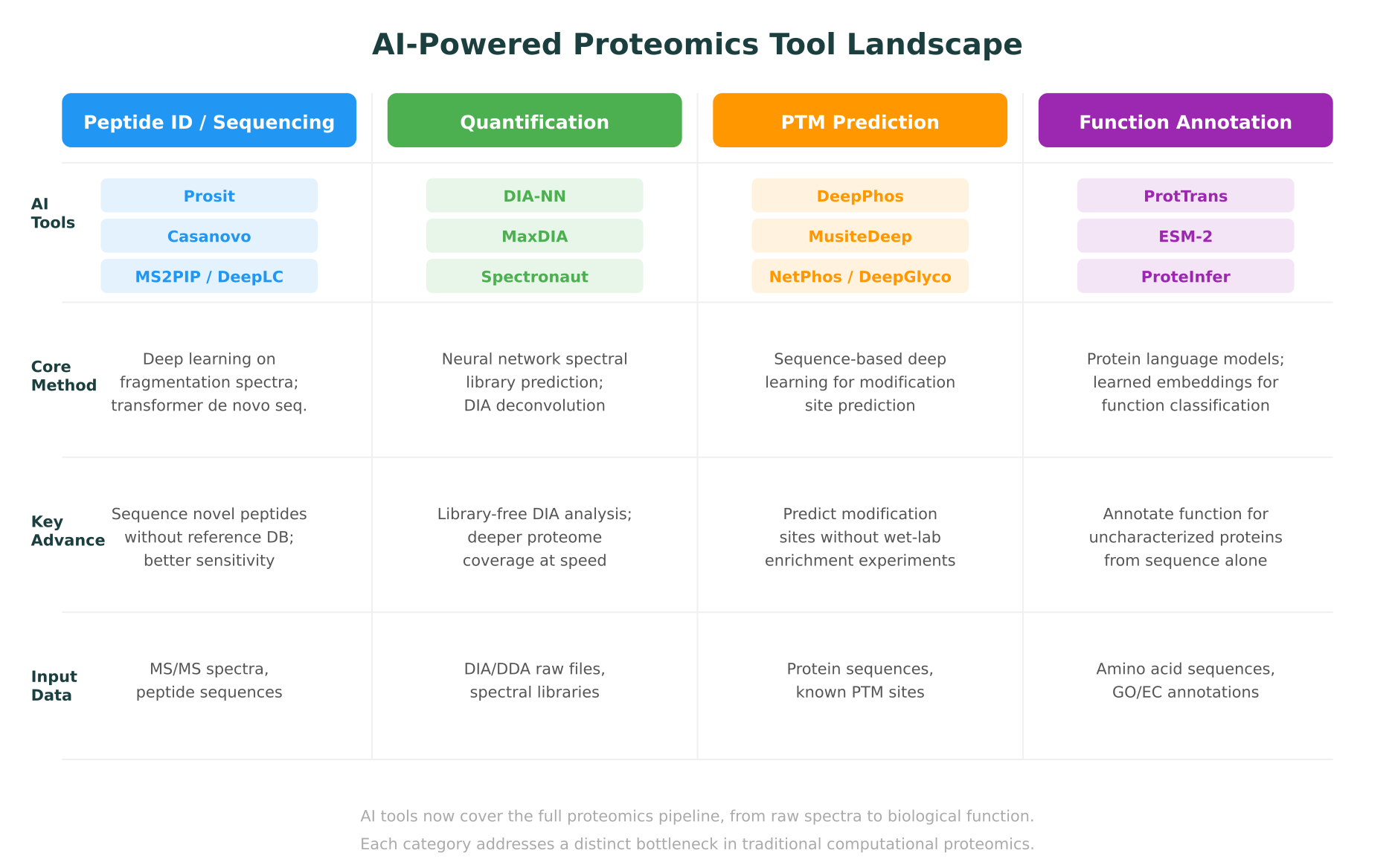
AI-Driven Protein Quantification
Identifying which proteins are present is only half the challenge. Measuring how much of each protein is present, and how that abundance changes between conditions, is the core quantitative question in proteomics.
The shift to data-independent acquisition
The proteomics field has been moving from data-dependent acquisition (DDA), where the mass spectrometer selects individual precursor ions for fragmentation, to data-independent acquisition (DIA), where all ions within a mass range are fragmented simultaneously. DIA generates more comprehensive data but produces highly complex, multiplexed spectra that are far harder to deconvolve. This is where AI has had perhaps its greatest quantitative impact.
DIA-NN: neural networks for label-free quantification
DIA-NN, developed by Vadim Demichev and published in Nature Methods (2020), uses deep neural networks to process DIA data without requiring pre-built spectral libraries. The software predicts spectral libraries in silico from protein sequence databases, then uses these predicted libraries alongside neural network-based signal extraction to identify and quantify proteins directly from DIA raw files.
The results are striking. In head-to-head comparisons, DIA-NN consistently identifies and quantifies more proteins from the same raw data than established tools like OpenSWATH or Spectronaut. On large-scale benchmarks, DIA-NN has demonstrated the ability to quantify over 10,000 proteins in a single human cell line experiment, approaching the estimated size of the expressed proteome. A 2024 study in Nature Biotechnology used DIA-NN to profile the proteomes of 180 cancer cell lines, generating one of the deepest pan-cancer proteomic maps to date.
MaxDIA, an extension of the widely used MaxQuant software, integrates deep learning-based spectral prediction into the MaxQuant ecosystem. Published in Nature Biotechnology in 2022, MaxDIA provides library-free DIA analysis with the familiar MaxQuant interface, lowering the barrier for labs already using the MaxQuant ecosystem for DDA data.
Single-cell proteomics
AI is also enabling the emerging field of single-cell proteomics. Technologies like SCoPE2 (Single Cell ProtEomics by Mass Spectrometry) and the more recent plexDIA approach generate extremely sparse data, where each cell contributes only a few thousand peptide identifications. AI-based imputation methods, trained on large bulk proteomics datasets, can fill in missing values and improve quantitative accuracy in single-cell experiments. A 2025 study in Cell Systems demonstrated that transformer-based imputation models reduced the missing value rate in single-cell proteomics data by over 50% while maintaining quantitative accuracy.
Post-Translational Modification Prediction
Proteins are not static sequences. After translation, they are chemically modified in ways that regulate their activity, localization, interactions, and degradation. Over 400 types of post-translational modifications (PTMs) have been described, with phosphorylation, ubiquitination, acetylation, methylation, and glycosylation being among the most studied. Experimentally, PTMs are detected through enrichment-based mass spectrometry workflows (such as TiO2 enrichment for phosphopeptides), but these experiments are expensive, technically demanding, and biased toward abundant modifications.
AI-based PTM prediction tools offer a complementary approach: predicting modification sites directly from protein sequences, without requiring enrichment experiments.
Phosphorylation prediction
DeepPhos, published in Bioinformatics (2019), uses a deep convolutional neural network to predict phosphorylation sites from protein sequences. Trained on experimentally validated phosphosites from PhosphoSitePlus, DeepPhos takes a window of amino acids centered on a potential modification site and predicts the probability that the serine, threonine, or tyrosine residue is phosphorylated. In benchmarks, DeepPhos outperformed earlier methods like NetPhos and GPS on independent test sets.
MusiteDeep, published in Briefings in Bioinformatics (2020), extends this approach to multiple PTM types simultaneously. Using an attention-based deep learning architecture, MusiteDeep predicts phosphorylation, ubiquitination, acetylation, methylation, SUMOylation, and other modifications from sequence alone. The attention mechanism provides interpretability by highlighting which sequence features the model considers most important for each prediction.
Glycosylation and beyond
Glycosylation, the attachment of sugar moieties to proteins, is one of the most complex and heterogeneous PTMs. DeepGlyco, published in Nature Communications (2022), uses deep learning to predict glycopeptide spectra, enabling more accurate identification of glycosylated peptides from mass spectrometry data. This is particularly relevant for therapeutic protein characterization, where glycosylation patterns affect drug efficacy and immunogenicity.
The practical value of PTM prediction
For computational proteomics researchers, AI-based PTM prediction serves several purposes. It generates hypotheses about which sites to target in enrichment experiments, prioritizing expensive wet-lab work. It provides functional context for protein sequences with no experimental PTM data. And when combined with structural information, predicting which residues are modified, and where those residues sit in the protein’s 3D structure, can illuminate regulatory mechanisms. A phosphorylation site in a disordered loop has different implications than one buried in a protein-protein interaction interface.
This is where the connection to structural biology tools becomes relevant. Platforms that integrate PTM prediction with protein structure visualization and mutation impact analysis can help researchers reason about the functional consequences of predicted modifications in three-dimensional context.
Protein Function Prediction with Language Models
Perhaps the most transformative application of AI in proteomics is protein function prediction using protein language models (PLMs). These models, trained on tens of millions of protein sequences using self-supervised learning (analogous to how large language models learn from text), generate dense numerical representations (embeddings) that capture evolutionary, structural, and functional properties of proteins.
The protein language model landscape
ESM-2, developed by Meta AI and published in Science (2023), is a 15-billion-parameter transformer trained on 65 million protein sequences from UniRef. ESM-2 embeddings encode rich biological information: proteins with similar functions cluster together in embedding space, even when they share little sequence identity. The model’s internal representations have been shown to capture secondary structure, contact maps, binding sites, and functional annotations with remarkable fidelity.
ProtTrans, published in IEEE Transactions on Pattern Analysis and Machine Intelligence (2022), is a family of protein language models based on architectures including BERT, Albert, and T5. Trained on UniRef and BFD (Big Fantastic Database, containing over 2 billion sequences), ProtTrans models provide per-residue and per-protein embeddings that serve as input features for downstream prediction tasks: subcellular localization, membrane topology, secondary structure, disorder prediction, and Gene Ontology (GO) term prediction.
ProteInfer, published by Google Research in eLife (2023), uses a dilated convolutional neural network to predict GO terms and EC (Enzyme Commission) numbers directly from protein sequences. Unlike methods that rely on sequence homology (like BLAST or InterProScan), ProteInfer can assign function to proteins with no detectable homologs, making it particularly valuable for metagenomic and orphan protein annotation.
Why PLM-based function prediction matters for proteomics
The practical significance for proteomics is substantial. In a typical proteomics experiment, 20-30% of identified proteins may lack functional annotation in databases like UniProt. For non-model organisms or metaproteomic samples, this fraction can exceed 50%. PLM-based function prediction fills this gap by assigning GO terms, predicting subcellular localization, and identifying potential enzymatic activities for proteins that would otherwise be labeled “uncharacterized.”
A 2024 study in Nature Methods compared PLM-based function prediction against the Critical Assessment of Functional Annotation (CAFA) benchmark and found that methods leveraging ESM-2 embeddings ranked among the top performers, outperforming homology-based methods for proteins in the “twilight zone” of sequence similarity (below 30% identity).
For researchers working at the intersection of proteomics and structural biology, PLM embeddings also provide a bridge to structure prediction. ESM-2’s internal representations have been used to predict protein structures (ESMFold), and the same embeddings that predict function can inform structural analysis. This connects directly to the broader ecosystem of AI-powered structure prediction tools that are reshaping how researchers think about protein biology.
Multi-Omics Integration: Connecting Proteomics to Genomics and Transcriptomics
AI proteomics tools are powerful on their own, but their value multiplies when proteomics data is integrated with genomic and transcriptomic information. This is the multi-omics perspective, and it addresses a fundamental biological reality: a genetic variant’s impact is ultimately measured at the protein level, but understanding the full mechanism requires tracing the path from DNA through RNA to protein.
The correlation problem
One of the most consistent findings in multi-omics research is that mRNA and protein levels correlate only moderately. Across large-scale studies, the Pearson correlation between mRNA expression and protein abundance in matched samples typically falls between 0.40 and 0.60. This means that roughly half of the variance in protein levels is not explained by transcript levels alone. Post-transcriptional regulation, translation efficiency, protein stability, and degradation all contribute to this disconnect.
For researchers, this has a practical implication: you cannot fully understand the proteome by measuring only the transcriptome. Proteomics data provides a distinct and necessary layer of biological information, as discussed in detail in our comparison of genomics, proteomics, and transcriptomics.
AI approaches to multi-omics integration
Several AI frameworks have been developed specifically for integrating proteomic data with other omics layers.
MOFA+ (Multi-Omics Factor Analysis), published in Genome Biology (2020), uses a Bayesian factor model to identify shared and data-type-specific sources of variation across omics layers. Given matched genomic, transcriptomic, and proteomic data from the same samples, MOFA+ identifies latent factors that explain coordinated changes across layers, revealing regulatory relationships invisible in single-omics analysis.
DeepOmix and similar deep learning frameworks use autoencoders or graph neural networks to learn joint representations from multiple omics data types. These models can predict patient outcomes or drug responses by integrating proteomic, transcriptomic, and genomic features in a way that captures cross-layer interactions.
A 2025 study in Cancer Cell applied multi-omics integration (genomics + transcriptomics + proteomics + phosphoproteomics) to 1,000 breast cancer tumors and identified proteomic subtypes that predicted treatment response more accurately than genomic or transcriptomic subtypes alone. The proteomics data revealed signaling pathway activation states that were invisible at the transcript level, directly informing treatment selection.
From variant to protein impact
For clinical and translational researchers, the most compelling application of multi-omics integration is connecting genetic variants to their protein-level consequences. A missense variant identified by whole-exome sequencing may or may not affect protein function. To assess its impact, you need to consider:
- Is the gene expressed? Transcriptomic data confirms whether the gene is active in the relevant tissue.
- Is the protein present? Proteomic data confirms whether the protein is actually produced and at what abundance.
- Is the protein modified? Phosphoproteomic or other PTM data reveals whether the variant affects a regulatory modification site.
- Does the variant affect protein structure? Structure prediction and stability analysis (DynaMut2, AlphaFold) predict whether the amino acid change destabilizes the fold or disrupts a functional interface.
- Is the variant in a functionally important region? Domain annotations, conservation scores, and 3D structure visualization contextualize the variant’s position.
This multi-layered analysis is exactly the kind of cross-domain reasoning that benefits from having all data types accessible in a single workspace, rather than switching between disconnected tools for each evidence type.
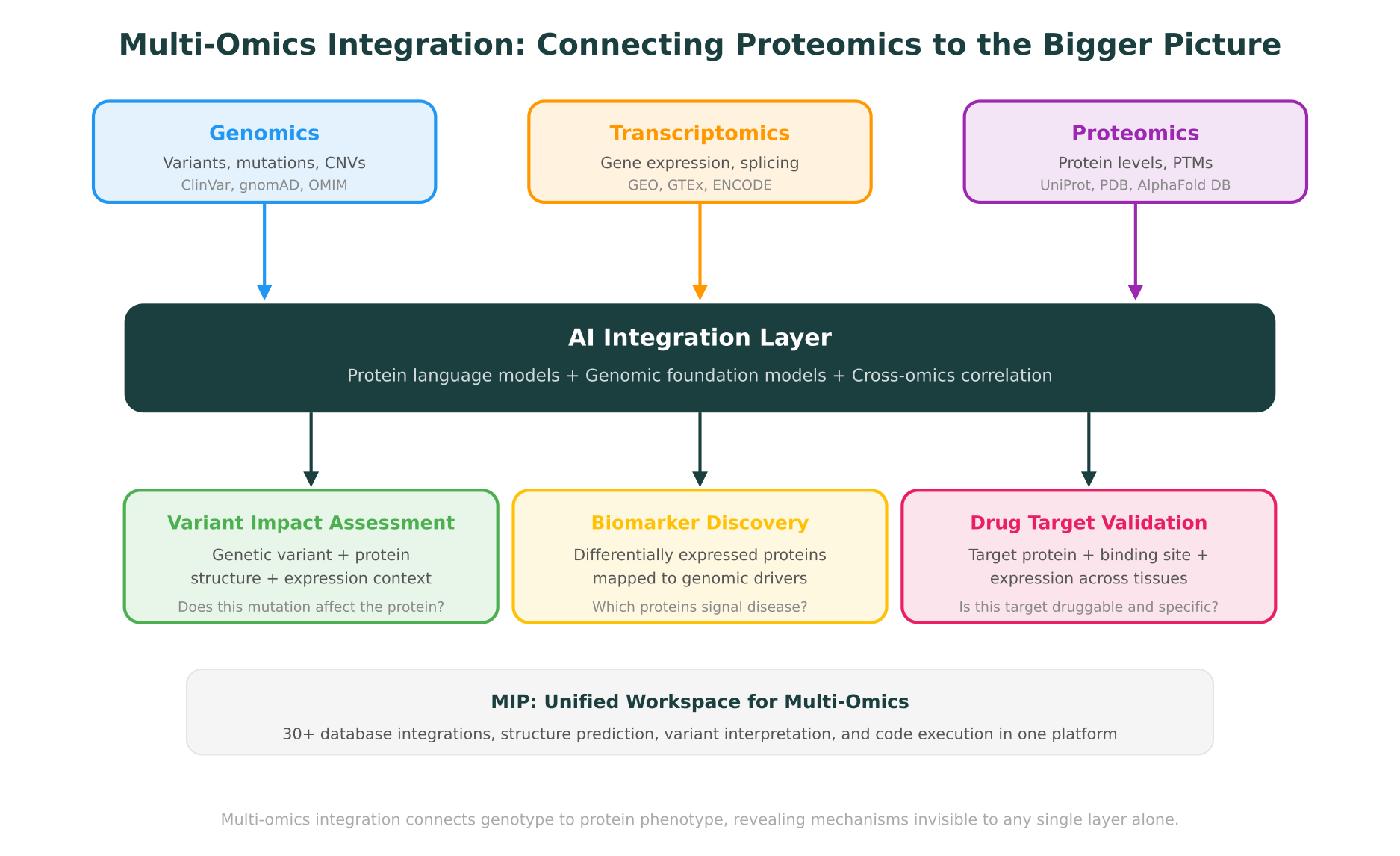
How MIP Supports Proteomic Context in Multi-Omics Research
Purna AI’s Molecular Intelligence Platform (MIP) was designed for the kind of cross-domain reasoning that modern proteomics demands. While MIP is not a mass spectrometry data processing tool, it provides the connective infrastructure that turns proteomics results into biological understanding.
Database integrations for proteomic context
MIP integrates with 30+ biological databases, including several that are directly relevant to proteomics workflows:
- UniProt: protein sequence, function annotation, post-translational modification data, subcellular localization, and protein-protein interaction information
- PDB (Protein Data Bank): experimentally determined protein structures for visualizing mutation sites and binding interfaces
- AlphaFold Database: predicted structures for proteins without experimental structures, covering over 200 million entries
- PharmGKB: pharmacogenomic relationships connecting genetic variants to drug response, including protein-level mechanisms
- ClinVar and gnomAD: variant frequency and clinical significance data that connects genetic findings to proteomic observations
When a researcher identifies a differentially expressed protein in their proteomics experiment, MIP enables them to immediately query its known variants, view its 3D structure in the integrated Molstar viewer, run stability analysis with DynaMut2, and check clinical databases for relevant associations, all through a natural language interface without switching tools.
Structure prediction and variant interpretation
For proteomic findings that implicate specific proteins, MIP’s structure prediction and variant interpretation capabilities provide immediate context. If a proteomics experiment reveals that a particular protein is differentially phosphorylated in disease samples, a researcher can use MIP to:
- Retrieve the protein structure from PDB or AlphaFold
- Visualize the phosphorylation site in 3D to assess its structural context
- Check whether the modification site overlaps with known functional domains
- Run DynaMut2 stability analysis if the site is near a disease-associated variant
- Query ClinVar and OMIM for clinically relevant connections
This workflow connects proteomics findings to structural and genomic evidence in a single session, the kind of cross-layer reasoning that molecular intelligence as infrastructure is designed to support.
Code execution for custom analysis
MIP’s containerized code execution environment allows researchers to run custom bioinformatics analyses directly in the platform. For proteomics researchers, this means running statistical analyses on quantification results, generating volcano plots, performing pathway enrichment, or building custom machine learning models on proteomic features, all without leaving the workspace where they access database information and structural tools.
What Comes Next for AI in Proteomics
The trajectory of AI in proteomics points toward several developments that are likely to materialize in the near term.
Multimodal foundation models that jointly model mass spectrometry data, protein sequences, and molecular structures are in active development at several research groups. These models promise to unify spectrum prediction, de novo sequencing, and function annotation into a single framework, rather than treating them as separate problems with separate tools.
Spatial proteomics is following the path of spatial transcriptomics. Technologies like MALDI imaging mass spectrometry and multiplexed ion beam imaging (MIBI) are generating spatially resolved protein abundance data from tissue sections. AI methods for integrating spatial proteomic data with spatial transcriptomics and histopathology imaging are emerging, with early publications in Nature Methods (2025) demonstrating proof of concept.
Clinical proteomics at scale is becoming feasible. The combination of rapid DIA methods (achieving full proteome coverage in under 30 minutes per sample) with AI-powered analysis tools like DIA-NN is making it practical to run proteomic profiling on thousands of clinical samples. Several large-scale clinical proteomics studies, including the UK Biobank Pharma Proteomics Project (over 50,000 participants), are now generating datasets that will serve as training data for the next generation of AI models.
Transfer learning from protein language models to proteomics-specific tasks is expanding. Rather than training de novo models for each application, researchers are fine-tuning general-purpose PLMs like ESM-2 on proteomics-specific datasets (phosphosite prediction, protein turnover prediction, protein-protein interaction prediction). This approach reduces the amount of task-specific training data needed and leverages the biological knowledge encoded in the pre-trained model.
Connecting AI Proteomics to the Broader Molecular Intelligence Ecosystem
AI in proteomics does not exist in isolation. The same protein language models that predict function from sequence also power protein folding and generative design. The same structural tools that contextualize proteomic findings also support drug target identification and docking. And the multi-omics integration frameworks that connect proteomics to genomics are the same ones advancing drug discovery.
For proteomics researchers and computational biologists, the practical takeaway is that the field’s AI tools are rapidly maturing. Spectrum prediction, de novo sequencing, DIA quantification, PTM prediction, and function annotation have all reached a level where they meaningfully outperform traditional computational approaches. The remaining challenge is integration: bringing these capabilities together with genomic, transcriptomic, and structural data in workflows that are accessible to biology teams, not just bioinformatics specialists.
This is the problem that molecular intelligence platforms are designed to solve. Not by replacing specialized proteomics software, but by providing the connective layer that links proteomic discoveries to the broader landscape of biological knowledge.
MIP is Purna AI’s Molecular Intelligence Platform, an AI-powered workspace for biology teams. Protein structure prediction, variant interpretation, 30+ database integrations, and code execution in one place. Explore the platform at purna.ai. Researchers can apply for up to $10,000 in free credits to run their analyses on MIP.
Explore Purna's Molecular Intelligence Platform
AI-powered workspace for biology teams to accelerate drug discovery from target identification to lead optimization.
Try Purna AI →
